Federico Perfetto, MD, PhD Dipartimento di Medicina Sperimentale e Clinica Università degli Studi di Firenze, CRR Amiloidosi AOU Careggi.
Francesco Cappelli; Ricercatore a tempo determinato AOU Careggi, Firenze.
Alessandro Barilaro, Dirigente Medico di Neurologia, Dipartimento Neuromuscoloscheletrico e Organi di Senso AOU Careggi Firenze.
L‘amiloidosi ereditaria da transtiretina (hATTR) fa parte di un gruppo di malattie rare e invalidanti caratterizzate dall’accumulo extracellulare, in vari tessuti, di materiale fibrillare insolubile costituito da proteine mal ripiegate in un processo noto come misfolding proteico. La malattia è determinata da mutazioni patogenetiche puntiformi in 3 dei 4 esoni da cui è costituito il gene della transtiretina (TTR) situato sull’autosoma 18. Si trasmette con modalità autosomica dominante e presenta una penetranza incompleta. La TTR è sintetizzata per circa il 90% dal fegato con piccole quantità prodotte anche dai plessi coroidei, dalla retina e dal pancreas. E’ una delle tre proteine che trasportano in circolo gli ormoni tiroidei e che veicola, in un complesso ternario, anche la retinol binding protein 4 e il retinolo. La TTR ha struttura quaternaria formata da 4 monomeri tutti uguali che individuano al loro interno una cavità legante soprattutto la fT4. Si tratta di una proteina instabile che tende naturalmente a dissociarsi in dimeri e monomeri soprattutto quando si trova in forma libera (80-85%). La presenza di alcune mutazioni genetiche amplifica enormemente questa instabilità e, pur non interferendo sulla funzione della proteina, ne provoca un progressivo misfolding con formazione di composti intermedi dotati di proteo tossicità cellulare e formazione di strutture fibrillari amiloidi che si depositano progressivamente nei tessuti. Ad oggi sono note oltre 130 varianti amiloidogeniche di TTR che mostrano una discreta correlazione tra genotipo e fenotipo clinico: alcune varianti si associano a un prevalente interessamento del sistema nervoso periferico e autonomo (fenotipo neurologico), altre si caratterizzano per un pressoché esclusivo danno cardiaco (fenotipo cardiaco) mentre in molti altri casi il coinvolgimento neurologico e quello cardiaco sono variamente associati (fenotipo misto). Nel mondo la mutazione Val50Met è la più frequente e risulta endemica nella popolazione portoghese dove determina una polineuropatia sensitiva motoria nota anche come Polineuropatia Amiloide Familiare (FAP) ad esordio precoce (<50 anni). La variante Val144Ile è invece la forma con fenotipo cardiaco più frequente nel mondo, in particolare negli Stati Uniti e nelle isole Caraibiche, nella popolazione di origine africana dove si manifesta, nei soggetti sopra i 60 anni, con insufficienza cardiaca rapidamente progressiva (Cardiopatia Amiloide Familiare, FAC). L’Italia non è un paese endemico per la hATTR anche se alcuni clusters regionali sono stati recentemente evidenziati ed indicati nella Figura 1.
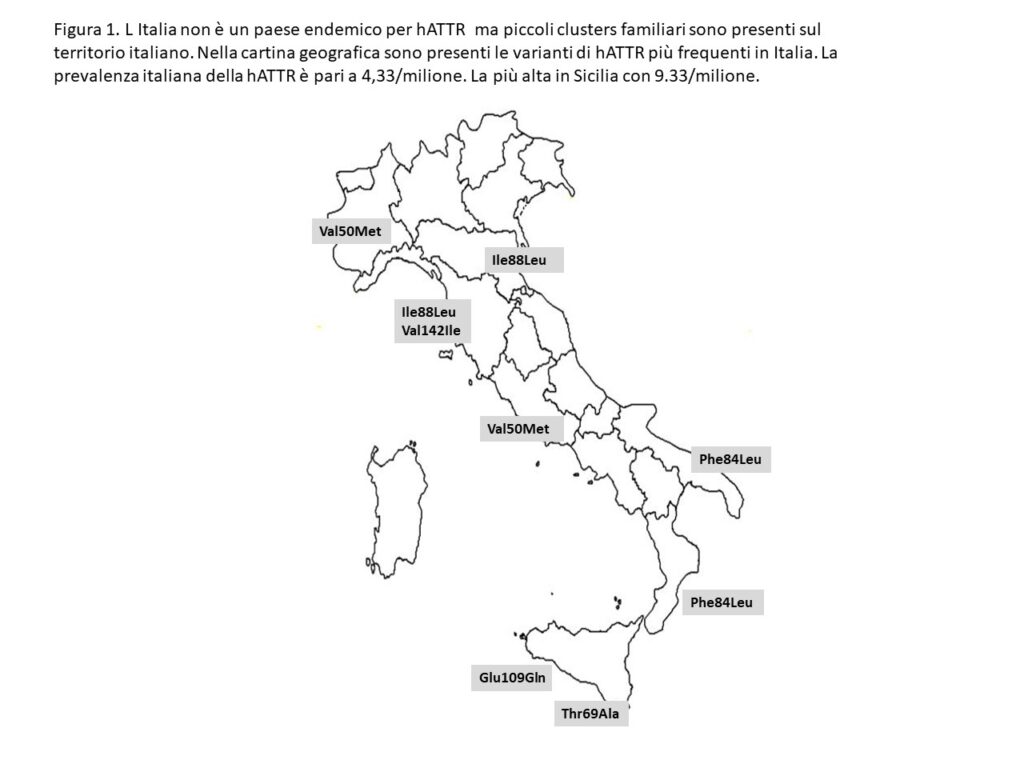
In Toscana sono particolarmente rappresentate 2 mutazioni con fenotipo cardiaco la Val144Ile e soprattutto la Ile88Leu con quest’ultima che si riscontra prevalentemente nei comuni a cavallo tra l’Appennino Toscano e Emiliano-Romagnolo. La prevalenza italiana della hATTR è pari a 4,33/milione di abitanti mentre quella Toscana è leggermente superiore, pari a 5,6/ milione di abitanti.
Le manifestazioni cliniche della hTTR sono variabili e dipendono dagli organi coinvolti, dal grado di infiltrazione e dal tipo di mutazione genetica. Mentre il coinvolgimento neurologico peggiora progressivamente la qualità di vita, l’interessamento cardiaco è il fattore prognostico negativo più importante riducendo, se non trattato, l’aspettativa di vita a pochi anni. L’infiltrazione miocardica causa ispessimento e rigidità delle pareti miocardiche, infiltra le valvole rendendole disfunzionanti e il pericardio. La compliance ventricolare è precocemente ridotta con conseguente aumento delle pressioni di riempimento telediastoliche, che si ripercuotono sugli atri causandone la dilatazione e successivamente congestione e ipertensione polmonare. La malattia può esordire con dispnea da sforzo, edemi declivi, epatomegalia e turgore giugulare, astenia ingravescente, progressiva intolleranza a una precedente terapia antipertensiva, sincope, bradiaritmie o fibrillazione atriale. L’elettrocardiogramma e l’ecocardiogramma mostrano alterazioni caratteristiche che permetto di confermare il sospetto clinico così come la RM cardiaca con mdc (vedi tabella 1).

La metodica di imaging che consente in molti casi di identificare con elevata sensibilità e specificità i depositi di amiloide da TTR nel cuore è la scintigrafia con tracciante osseo (99mTc-DPD, 99mTc-HMDP e 99mTc-PYP) che mostra una inattesa captazione del tracciante a livello miocardico a scapito dell’osso. L’interessamento del sistema nervoso periferico determina una polineuropatia assonale sensitivo-motoria, simmetrica, con distribuzione lunghezza-dipendente, che solitamente esordisce agli arti inferiori. Inizialmente sono colpite soprattutto le fibre nervose di piccolo calibro, causando dolore neuropatico, parestesie e alterazione della sensibilità termica. Gradualmente il danno progredisce anche a carico delle fibre nervose di maggiore calibro, sia sensitive sia motorie e si estende anche agli arti superiori. Contemporaneo e grave è anche il coinvolgimento del sistema nervoso autonomo (SNA) che determina disfunzione erettile, ipotensione ortostatica, ritenzione urinaria e turbe della motilità gastrointestinale con importante perdita di peso e malnutrizione. Prototipo di questa forma è la variante Val50Met ad esordio precoce, endemica soprattutto nel nord del Portogallo e in alcune zone del Giappone e della Svezia. In Italia la Val50Met ha un esordio più tardivo (>50 anni), mostra un coinvolgimento del SNA minore, una maggiore compromissione delle fibre motorie e soprattutto un precoce e maggiore coinvolgimento cardiaco che la rendono il prototipo delle forme a fenotipo misto. Raramente e in alcune particolari mutazioni sono coinvolti il sistema nervoso centrale con infiltrazione dei vasi leptomeningeali con sintomatologia che ricorda quadri ischemici atipici e/o simil epilettici e gli occhi con precoce distacco di vitreo e progressiva perdita del visus. Infine in una discreta percentuale di pazienti manifestazioni muscolo scheletriche quali la sindrome del tunnel carpale o la stenosi del canale lombare in cui sono dimostrabili depositi di amiloide TTR correlata, precedono di alcuni anni le tipiche manifestazioni neurologiche e cardiache.
La conoscenza della malattia, un alto indice di sospetto diagnostico, la capacità di collegare tra loro sintomatologie diverse e apparentemente distanti, sono fondamentali per una diagnosi precoce di questa rara malattia. La storia familiare di questi pazienti è quasi sempre muta ma la presenza di malattie cardiache o neurologiche ad eziologia ignota negli antenati o nei fratelli deve essere attentamente ricercata. Le caratteristiche cliniche particolari dello scompenso cardiaco nei pazienti con hATTR con quadri ECG/ecocardiografici peculiari, l’associazione a quadri sfumati di polineuropatia e disautonomia o viceversa una polineuropatia con disautonomia in un paziente con edemi periferici e fibrillazione atriale devono far sospettare una possibile amiloidosi da TTR soprattutto se il paziente ha origine da alcune particolari regioni della nostra penisola. Molte volte i pazienti con hATTR ricevono diagnosi errate che vanno dalla cardiomiopatia ipertrofica, alla cardiopatia ipertensiva o ischemica oppure, se il quadro clinico dominante è neurologico, dalla polineuropatia diabetica alla SLA, ad una polineuropatia demielinizzante infiammatoria cronica (CIPD). I pazienti in cui la neuropatia non risponde alla terapia specifica o che presentano una sproporzionata componente disautonomica o in quei pazienti in cui l’entità della malattia diabetica non giustifica la presenza di una grave neuropatia periferica, devono essere riconsiderati includendo anche la hATTR tra le possibili diagnosi differenziali. I pazienti con sospetta hATTR vanno riferiti allo specialista cardiologo o neurologo, meglio se afferenti ad un centro esperto in questa patologia. La diagnosi, che deve obbligatoriamente includere la ricerca di una mutazione di significato patologico del gene TTR, può essere confermata sia in modo invasivo che non invasivo. Nei casi di pazienti con coinvolgimento cardiologico, una volta esclusa una forma AL mediante la dimostrazione della assenza di una componente monoclonale nel siero e/o nelle urine, la presenza di una captazione miocardica significativa (grado Perugini 2 o 3) alla scintigrafia con tracciante osseo, è oggi sufficiente come evidenza indiretta del deposito di TTR a livello cardiaco e può pertanto sostituire una biopsia positiva. Nei casi con componente monoclonale presente o nei casi con assenza (grado Perugini 0) o bassa captazione cardiaca alla scintigrafia (grado Perugini 1) la biopsia è obbligatoria. Nei pazienti a fenotipo cardiaco la biopsia endomiocardica è la metodica preferita mentre nelle forme puramente neurologiche le sedi prescelte sono la biopsia chirurgica del grasso sottocutaneo periombelicale, la biopsia delle ghiandole salivari minori e per ultima la biopsia del nervo che viene eseguita sempre meno frequentemente per l’invasività e gli esiti che conseguono alla sua esecuzione. La presenza di amiloide sul materiale biopsiato sarà rivelata dalla caratteristica birifrangenza verde brillante al microscopio polarizzatore dopo colorazione con Rosso Congo. Sui depositi si deve inoltre procedere alla loro tipizzazione al fine di confermare definitivamente la presenza di depositi di TTR.
La strategia terapeutica dei pazienti con hATTR si basa su due cardini: la terapia di supporto del paziente al contempo cardiopatico e neuropatico e la terapia specifica della hATTR volta ad arrestare la deposizione di amiloide nei tessuti bersaglio. A tal fine la collaborazione tra cardiologi, neurologi, terapisti del dolore, nutrizionisti, psicologi, gastroenterologi è di fondamentale importanza per il paziente e la sua famiglia. La terapia specifica della hATTR ha subito un cambiamento epocale nel 2018 con la pubblicazione di alcuni studi fondamentali che hanno dimostrato come la strategia del gene silencing e della ricerca di molecole stabilizzanti la TTR siano in grado di modificare la storia naturale della malattia. Il tafamidis, molecola in grado di ridurre il misfolding della TTR, dopo 30 mesi di terapia in pazienti con ATTRwt o hATTR con fenotipo cardiologico, ha ridotto significativamente il rischio di morte e di ospedalizzazioni per cause cardiovascolari rispetto al gruppo placebo. Sulla base di questi dati il trattamento con tafamidis al dosaggio di 80 mg/die è stato approvato da AIFA per la cardiomiopatia da transtiretina.
Ancora più interessanti sono stati i risultati con i farmaci silenziatori genici, Inotersen e Patisiran. In particolare quest’ultimo che, sfruttando il processo di RNA interference che ha permesso di sopprimere fino ad oltre l ‘80% la produzione epatica di ATTR normale e mutata, ha mostrato la capacità di indurre un significativo miglioramento della polineuropatia periferica e autonomica nei pazienti con hATTR con neuropatia di grado I e II rispetto al placebo, preservando sia la qualità di vita che la capacità funzionale dei pazienti con un ottimo profilo di sicurezza. Il farmaco, che si somministra ogni tre settimane per via endovenosa, preceduto da una premedicazione, è stato approvato dall’AIFA con indicazioni per pazienti con hATTR con neuropatia in stadio I e II. Recentemente una nuova molecola, Vutrisiran, che condivide con Patisiran lo stesso meccanismo d’azione, si è dimostrata altrettanto se non più efficace nel ridurre i livelli circolanti di TTR, con il vantaggio di poter essere somministrato per via sottocutanea e di possedere una maggiore durata di azione, che ha permesso di allungare gli intervalli di somministrazione a tre mesi senza necessità di premedicazione e di somministrazione ospedaliera. Anche questa terapia è stata recentemente approvata da AIFA con rimborsabilità per pazienti con hATTR e neuropatia stadio I e II.
In conclusione una malattia rara come la hATTR che fino ad alcuni anni fa era priva di efficaci strategie terapeutiche, grazie alle sempre maggiori capacità diagnostiche, alla migliore conoscenza dei meccanismi patogenetici e alla diffusione della sua conoscenza nel mondo medico è uscita dal novero delle malattie orfane trovando trattamenti innovativi ed efficaci per migliorarne il decorso e offrire un futuro migliore alla maggior parte di questi pazienti e alle loro famiglie.
federico.perfetto@unifi.it
L’autore Federico Perfetto, dichiara di aver ricevuto i seguenti finanziamenti o di avere i seguenti contratti in corso, personali o istituzionali, con soggetti pubblici o privati i cui prodotti o servizi sono citati nella pubblicazione: Advisory Board Pfizer, Alnylam Pharmaceuticals, Akcea Terapeutics.
L’autore Alessandro Barilaro, dichiara di aver ricevuto i seguenti finanziamenti o di avere i seguenti contratti in corso, personali o istituzionali, con soggetti pubblici o privati i cui prodotti o servizi sono citati nella pubblicazione: Personal fee Alnylam Pharmaceuticals.
L’ESPERIENZA TOSCANA DEL GRUPPO “NEURO – TTR” L’approccio multispecialistico a patologie sistemiche e complesse come l’amiloidosi appare di fondamentale importanza. Siamo infatti di fronte ad una patologia sistemica complessa che richiede la massima competenza di tutti gli specialisti coinvolti. Quando queste figure sono integrate, coordinate e ben affiatate rappresentano un valore aggiunto nella gestione della patologia, considerando che anche all’interno della stessa specialistica, la complessità della malattia può richiedere la collaborazione di più figure con diverse esperienze e competenze. Per questo un gruppo di neurologi toscani* che si occupa di neuropatia amiloidotica familiare, si è confrontato in modo pratico sul percorso diagnostico e terapeutico della patologia. Il gruppo, con competenze complementari, ha redatto un documento che fornisce indicazioni guida di buona prassi sul processo di diagnosi e la gestione dei pazienti e dei familiari portatori di mutazione. Tale gruppo regionale “Neuro-TTR” ha gettato le basi per continuare a lavorare in rete sia nella gestione dell’attività clinica sia per progetti di ricerca che possano migliorare il percorso fin qui realizzato. * AOU Careggi: Alessandro Barilaro, Riccardo Caramelli, Maria Lombardi AOU Pisana: Gabriele Siciliano, Erika Schirinzi AOU Senese: Federica Ginanneschi, Nila Volpi USL Toscana Centro: Maria Briccoli Bati, Roberto Fratangelo Medici in formazione specialistica Università degli studi di Firenze: Antonio Lotti, Carla Fasano, Ugo Mollo. |
