Giovanni Taccetti, Dirigente medico, Centro Regionale Toscano per la Fibrosi Cistica, Azienda Ospedaliero-Universitaria Meyer IRCCS
Michela Francalanci, Biologo, Study coordinator – Data manager Centro Regionale Toscano per la Fibrosi Cistica Azienda Ospedaliero-Universitaria Meyer IRCCS
Francesca Menegazzo, Dirigente medico, Direzione Sanitaria, Azienda Ospedaliero-Universitaria Meyer IRCCS
Abstract
La prognosi della fibrosi cistica, una grave malattia letale ereditaria che colpisce principalmente l’apparato respiratorio, ha tratto giovamento nel corso degli anni dal miglioramento complessivo delle cure e la maggior parte dei pazienti raggiunge ora l’età adulta. Recentemente la terapia è stata rivoluzionata da un nuovo tipo di trattamento che per la prima volta agisce sulle cause della malattia ed è efficace nel 75% degli affetti.
Parole chiave: Fibrosi Cistica; Screening; Malattie Rare; Cure Innovative; modulatori proteina CFTR;
La fibrosi cistica (FC) è la più frequente malattia genetica letale ereditaria dei caucasici. La sua incidenza in Toscana, calcolata in base ai dati del programma regionale di screening neonatale per la FC, attivo nella nostra Regione fin dal 1984, è di 1 nuovo affetto ogni 4000 neonati. La FC è una malattia multisistemica che interessa principalmente i polmoni, le vie aeree superiori, il pancreas, il fegato, l’intestino e l’apparato riproduttivo (principalmente i dotti deferenti). È caratterizzata dalla produzione di muco particolarmente denso a livello delle mucose e i pazienti sviluppano di solito, nel corso del tempo, una sintomatologia caratterizzata da malnutrizione e da infezioni polmonari batteriche recidivanti che determinano un’evoluzione verso l’insufficienza respiratoria.
La malattia, a trasmissione autosomica recessiva, è causata da undifetto su base genetica della proteina transmembrana CFTR (Cystic Fibrosis Transmembrane Conductance Regulator) che regola il movimento degli ioni (principalmente cloro e bicarbonato) attraverso le membrane delle cellule epiteliali. Un difetto nella sintesi e nel processamento di tale proteina determina anomalie delle secrezioni esocrine che diventano “disidratate” e dense, andando ad ostruire i dotti secretori delle ghiandole stesse. Come particolarità in questa malattia le ghiandole sudoripare producono un sudore con concentrazione di cloro molto alta e proprio questo motivo ha suggerito ai clinici di adottare il test del sudore come esame diagnostico.
La scoperta del gene CFTR, che codifica per la proteina omonima e la comprensione delle modalità con cui le diverse mutazioni genetiche ne determinano un’anomala funzione sono stati passi determinanti per conoscere in maniera più approfondita questa patologia e per rendere disponibile nel corso del tempo una terapia non più basata sul trattamento dei sintomi ma sulla causa eziologica. A oggi sono state identificate più di 2000 varianti del gene CFTR, di cui 360 sono classificate come sicuramente causanti la malattia. Tali varianti sono state categorizzate in diverse classi in base ai loro effetti sulla proteina CFTR in termini di produzione, processazione, migrazione in membrana, funzione e stabilità. La variante “causing” più frequente è la F508del, che si associata ad un fenotipo clinico severo con insufficienza del pancreas esocrino. In Italia è presente nel 75% circa dei cromosomi dei soggetti affetti.
Nella nostra Regione la diagnosi di fibrosi cistica viene di solito effettuata con accertamenti diagnostici in soggetti risultati positivi al programma di screening neonatale e si basa sulla positività del test del sudore e sull’analisi molecolare del gene CFTR.
Fino a pochi anni fa la terapia della FC aveva come obiettivo il trattamento sintomatico della malattia, principalmente basato sulla terapia delle esacerbazioni respiratorie e dell’insufficienza del pancreas esocrino. Nell’ultimo decennio, accanto ai notevoli progressi già compiuti nel trattamento dei sintomi della malattia, sono state individuate molecole in grado di correggere gli specifici tipi di malfunzionamento della proteina. Abbiamo quindi oggi a disposizione farmaci innovativi per un numero crescente di pazienti in base al loro esatto genotipo CFTR (terapia di precisione). Queste nuove molecole, note con il termine “modulatori della proteina CFTR”, costituiscono per la prima volta un trattamento che agisce sulla causa stessa della malattia e non sui suoi sintomi.
Oggi il 75% dei pazienti FC, in relazione al proprio genotipo CFTR, ha a disposizione farmaci in grado di ripristinare il funzionamento della proteina transmembrana. I modulatori della proteina CFTR sono associazioni di molecole con un effetto di correttore della proteina (favoriscono la maturazione della proteina F508del-CFTR e la sua permanenza sulla membrana) e di potenziatore dell’attività di canale transmembrana (stimolano l’attività della proteina presente in membrana, aprendo il canale e permettendo il passaggio degli ioni cloruro e bicarbonato). La sinergia di azione fra tale combinazione di molecole migliora gli esiti clinici della malattia poiché incrementa la funzionalità respiratoria dei pazienti e riduce il numero di esacerbazioni polmonari.
Il farmaco maggiormente efficace, di più recente introduzione, è una combinazione di 3 principi attivi, elexacaftor/tezacaftor/ivacaftor (ETI), che agiscono ripristinando la funzione di canale transmembrana in pazienti che presentino almeno una mutazione F508del. L’attuale autorizzazione all’immissione in commercio in Europa è stata supportata dai risultati positivi di due studi recentemente pubblicati. I due trial clinici, ai quali hanno preso parte anche pazienti in follow-up presso il Centro Regionale Toscano Fibrosi Cistica dell’AOU Meyer IRCCS, hanno valutato l’efficacia e la sicurezza di questo farmaco e hanno evidenziato un impatto positivo sulla malattia polmonare e sullo stato nutrizionale.
Presso il centro Centro Regionale Toscano FC AOU Meyer IRCCS, in collaborazione con AOU Careggi e con i Servizi di Supporto per la Fibrosi cistica di Livorno e Grosseto, sono seguiti più di 400 pazienti con diagnosi di Fibrosi Cistica (62% adulti e 38% sotto i 18 anni), con una casistica in costante aumento negli anni.
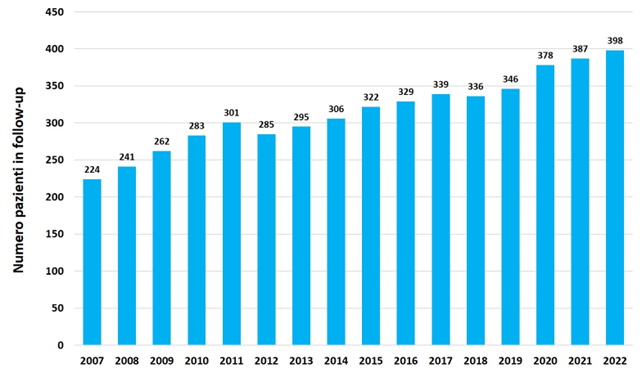
Figura 1. Centro Regionale Toscano FC AOU Meyer IRCCS: pazienti in follow-up
Dei pazienti seguiti al Centro Regionale Toscano FC sono note le mutazioni causanti malattia; i soggetti omozigoti F508del sono il 24.1% mentre gli eterozigoti composti con un allele F508del sono il 44.9%.
I pazienti con almeno una variante F508del rispondono alla nuova terapia con 3 principi attivi (ETI) che è stata recentemente approvata da AIFA per la commercializzazione in pazienti di età superiore ai 12 anni. A settembre 2022, essendo stata dimostrata la sicurezza della terapia, le indicazioni sono state estese ai pazienti dai 6 anni di età. Negli Stati Uniti d’America gli organismi regolatori locali (FDA) hanno già approvato la terapia anche per i pazienti dai 2 anni di età, mentre in Italia la procedura di approvazione da parte di AIFA è attualmente in corso.
Presso il Centro Regionale Toscano FC dell’AOU Meyer IRCCS sono a oggi in terapia con nuovi farmaci (ETI) 220 pazienti (140 pazienti adulti e 80 pediatrici), alcuni dei quali hanno inizialmente partecipato a programmi di uso compassionevole del farmaco e sono stati successivamente inseriti in uso commerciale. Progressivamente verranno inseriti in terapia tutti i pazienti in età pediatrica che soddisfino i requisiti che verranno stabiliti da AIFA per genotipo ed età.
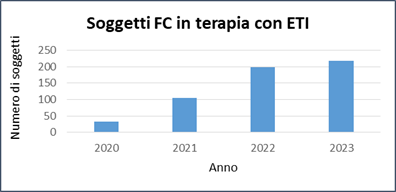
Figura 2. Incremento del numero di pazienti in terapia con Elexacaftor/Tezacaftor/Ivacaftor (ETI) presso il Centro Regionale Toscano FC dell’AOU Meyer IRCCS
Come da regolare pratica clinica, i pazienti continuano ad essere valutati ogni 3 mesi con monitoraggio dei dati auxometrici, della funzione respiratoria, del numero di riacutizzazioni e dell’uso di risorse terapeutiche. Poiché i nuovi farmaci vengono metabolizzati dal fegato, i pazienti in terapia sono inseriti in un rigoroso programma di controllo della funzione epatica con esami ematici ogni 3 mesi per valutare l’andamento degli indici epatici durante il primo anno di terapia e successivamente ogni 6-12 mesi. Inoltre periodicamente viene eseguito il test del sudore, i cui valori, diminuendo, testimoniano l’efficacia della terapia in atto. Ad ogni visita vengono inoltre valutate la situazione microbiologica delle vie aeree e la qualità di vita (tramite l’apposito score CFQ-R).
Tali dati sono richiesti da AIFA per il controllo dell’appropriatezza prescrittiva che avviene su un apposito registro di monitoraggio dei farmaci innovativi.
Le figure di seguito illustrano i miglioramenti registrati per i pazienti in terapia con ETI nel corso del tempo riguardo a:
– funzione respiratoria (FEV1%)
– stato nutrizionale (BMI)
– punteggio relativo alla sfera respiratoria del questionario sulla qualità di vita in Fibrosi Cistica (CFQ-R)
– valori di cloro sudorale (mmol/L)
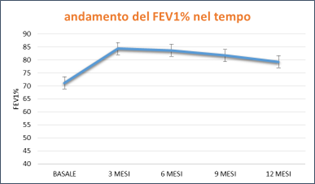
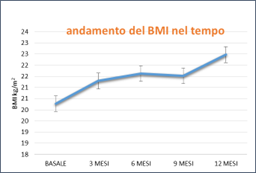
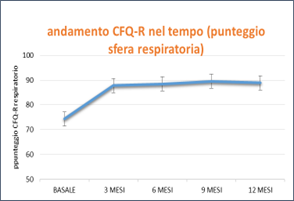
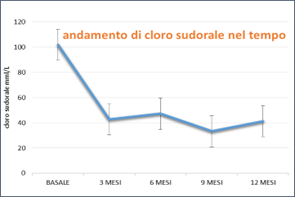
Figura 3. Andamento dei parameti oggetto di studio in pazienti FC in terapia con Elexacaftor/Tezacaftor/Ivacaftor (ETI) nel corso del tempo.
I pazienti FC in trattamento con ETI (i soggetti con almeno una variante F508del nel loro genotipo CFTR) mostrano un miglioramento della funzione polmonare (in termini di FEV1%) e dello stato nutrizionale (in termini di BMI), un incremento del punteggio dello score relativo alla qualità di vita (sfera respiratoria del CFQ-R) e una riduzione della concentrazione di cloro nel sudore fino a valori non più patologici. I miglioramenti ottenuti nel corso del tempo sono statisticamente significativi quando confrontati con i valori prima dell’inizio della terapia (basali). Inoltre tutti i parametri vanno incontro a miglioramento dopo solo 3 mesi di terapia e si mangono stabili nel tempo. Grazie alla terapia con i nuovi farmaci presso il Centro Regionale Toscano FC AOU Meyer IRCCS abbiamo ottenuto un miglioramento consistente delle condizioni cliniche dei pazienti, i cui valori osservati sono in accordo con le pubblicazioni internazionali che hanno portato all’autorizzazione e all’immissione in commercio delle nuove molecole in Europa.
Per il complessivo miglioramento delle condizioni cliniche si è parallelamente assistito nel corso del tempo ad una drastica riduzione del numero delle giornate di degenza in ricovero ordinario.
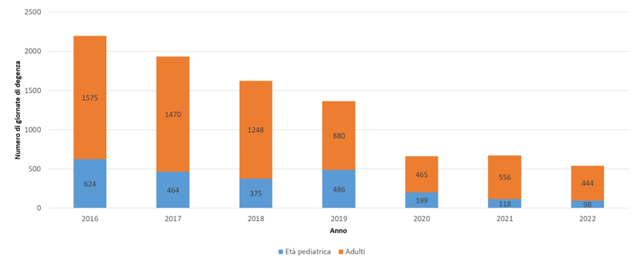
Figura 4. Giornate di ricovero ordinario in pazienti affetti da FC nel corso degli anni
Ulteriori aspettative sono ora riposte nella ricerca di strategie terapeutiche nei confronti di quei soggetti (attualmente il 25%) che per le loro caratteristiche genetiche (assenza di un allele F508del nel loro genotipo CFTR) non possono ancora trarre benefici dalla terapia con le nuove molecole che abbiamo oggi a disposizione.
In conclusione, nel corso del tempo le condizioni cliniche generali dei pazienti FC e la loro sopravvivenza sono drasticamente cambiate sia per l’ottimizzazione delle strategie terapeutiche già note che per l’uso dei nuovi farmaci. Quella che era fino a poco tempo fa la malattia letale genetica più comune dell’infazia si sta trasformando sempre più in una malattia cronica dell’adulto, con un miglioramento complessivo della qualità di vita delle persone affette.
Ringraziamenti: si ringraziano tutti gli operatori sanitari della Rete Regionale Toscana per la Fibrosi Cistica
