Ettore Giustini Saffi, medico già membro Commissione Terapeutica Reg. Toscana, Consulente AIFA
Le ADR (Adverse Drug Reactions) hanno un impatto significativo non solo da un punto di vista clinico ma anche da un punto di vista economico.
Uno studio fondamentale da questo punto di vista, effettuato su quasi 20.000 pazienti ricoverati in ospedale nel Regno Unito, ha rilevato che le ADR portano a una media di otto giorni aggiuntivi di degenza ospedaliera e sono associate a circa € 706 milioni/anno, comprese le ADR giudicate potenzialmente prevenibili. Una revisione sistematica più recente di 29 studi osservazionali ha messo in evidenza che il costo totale incrementale per ciascun paziente con un evento avverso in corso di terapia variava da circa € 702 a € 7,318 milioni/anno. Tuttavia questo studio non ha esaminato l’impatto economico delle ADR nello specifico, cioè di quelle reazioni sicuramente associate all’esposizione al farmaco.
Dato che le ADR sono molto comuni e che molte di queste possono essere prevenibili, è fondamentale comprendere e quantificarne l’onere economico in quanto è possibile ridurre sia i costi diretti che quelli indiretti associati, salvaguardando allo stesso tempo la salute dei pazienti. Lo scopo del presente studio è stato quindi svolgere una revisione sistematica degli studi osservazionali che hanno valutato l’impatto economico delle ADR prevenibili.
Per quanto riguarda l’analisi dei costi, tutti gli studi hanno valutato i costi diretti e solo 3 studi (16,7%) hanno valutato sia i costi diretti che indiretti.
Nel contesto ospedaliero, i costi diretti dovuti ad ADR prevenibili (per ospedalizzazione) variavano da un minimo di € 2.851 a un massimo di € 9.015 con un eccesso della durata del ricovero che andava da 4,2 a 13,0 giorni.
D’altra parte, negli studi condotti nel contesto ambulatoriale, i costi diretti associati ad ADR prevenibili andavano da un minimo di € 174 (costo medio di una visita in pronto soccorso) a un massimo di € 8.515 (costo medio ospedaliero per ammissione) con una LOS che andava da 7,0 a 9,3 giorni. Infine, negli studi condotti sia nel setting ospedaliero che ambulatoriale, i costi diretti dovuti ad ADR prevenibili variavano da un minimo di € 63,8 (per i farmaci prescritti) a un massimo di € 2.721 (per il ricovero).
L’eccesso della durata del ricovero era in media di 8,5±4,2 giorni. Tra i 3 studi che hanno valutato i costi diretti e indiretti, il primo ha stimato un costo sanitario indiretto medio di € 1.712 per ospedalizzazione per le persone di età inferiore ai 65 anni, tenendo conto dei costi di produttività come l’assenza dal lavoro e la riduzione della produttività sul lavoro. Inoltre, il costo totale della perdita di produttività per l’ospedalizzazione variava da € 61 per un uomo di 19 anni con degenza di 1 giorno a € 13.234 per un uomo di 37 anni per degenza di 38 giorni.
Il secondo ha calcolato il costo sanitario indiretto medio dei servizi gestiti da società esterne (come la lavanderia o il servizio di ristorazione) per gli accessi al pronto soccorso associati a una malattia iatrogena che si verifica tra pazienti non ospedalizzati od ospedalizzati. Questo ammontava rispettivamente a € 909 (±248) e € 78 (±62) €.
Infine, nel terzo studio, il costo indiretto medio della malattia per i pazienti con almeno un ADR prevenibile era pari a € 2.674 (IC al 95% 2.066-3.282).
Questo studio di popolazione1, che ha coinvolto adulti over 65 anni provenienti sia dal setting di cure primarie che specialistiche, ha mostrato come la spesa sanitaria degli individui che hanno manifestato reazioni avverse da farmaco fosse sproporzionatamente elevata (25% dei costi totali) rispetto alla prevalenza di tali reazioni avverse da farmaco nella popolazione in studio (8% degli 813 partecipanti).
Gli individui con prescrizioni potenzialmente inappropriate avevano una spesa sanitaria più elevata rispetto a quelli senza prescrizioni potenzialmente inappropriate (1958 € vs 881 €) e questa spesa era principalmente determinata dal costo degli interventi sanitari e non dal costo dei farmaci.
1 Spesa sanitaria dovuta a reazioni avverse da farmaci e a prescrizioni potenzialmente inappropriate negli anziani: uno studio basato sulla popolazione
Healthcare costs of adverse drug reactions and potentially inappropriate prescribing in older adults: a population-based study – Robinson EG, Hedna K, Hakkarainen KM, Gyllensten H. BMJ Open 2022; 12(9):e062589
Bibliografia
The economic burden of preventable adverse drug reactions: a systematic review of observational studies D Formica, J Sultana, PM Cutroneo, et al. Expert Opin Drug Saf 2018; 17:681-695.
Flash di Farmacovigilanza ( FV) nella storia
Nel 1922 fu indagato il salvarsan (composto arsenicale organico impiegato per la lue) imputato di provocare ittero.
Nel 1937 negli USA morirono 107 persone dopo aver assunto un elisir di sulfanilamide contenente il solvente glicol dietilenico i cui effetti tossici erano già noti, senza che il produttore ne fosse a conoscenza.
Da allora venne istituita la Food and Drug Administration (FDA) per indagare sulla sicurezza dei farmaci prima della loro immissione in commercio
Nonostante vari casi, verificatisi nella prima metà del XX secolo, l’opinione pubblica e le istituzioni rimasero abbastanza indifferenti ai problemi di FV fino al 1961.
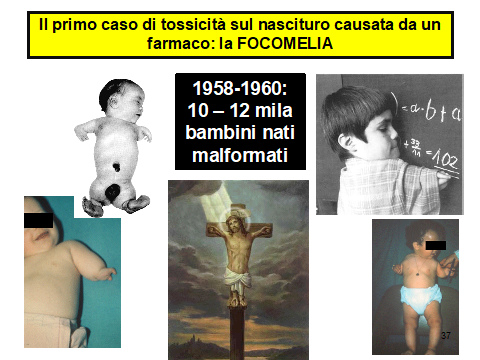
A dicembre di quell’anno The Lancet pubblicò una lettera del Dr. McBride che chiedeva se esisteva un nesso di causalità fra alterazioni di sviluppo degli arti (focomelia) di bambini nati da madri che avevano assunto in gravidanza la talidomide.
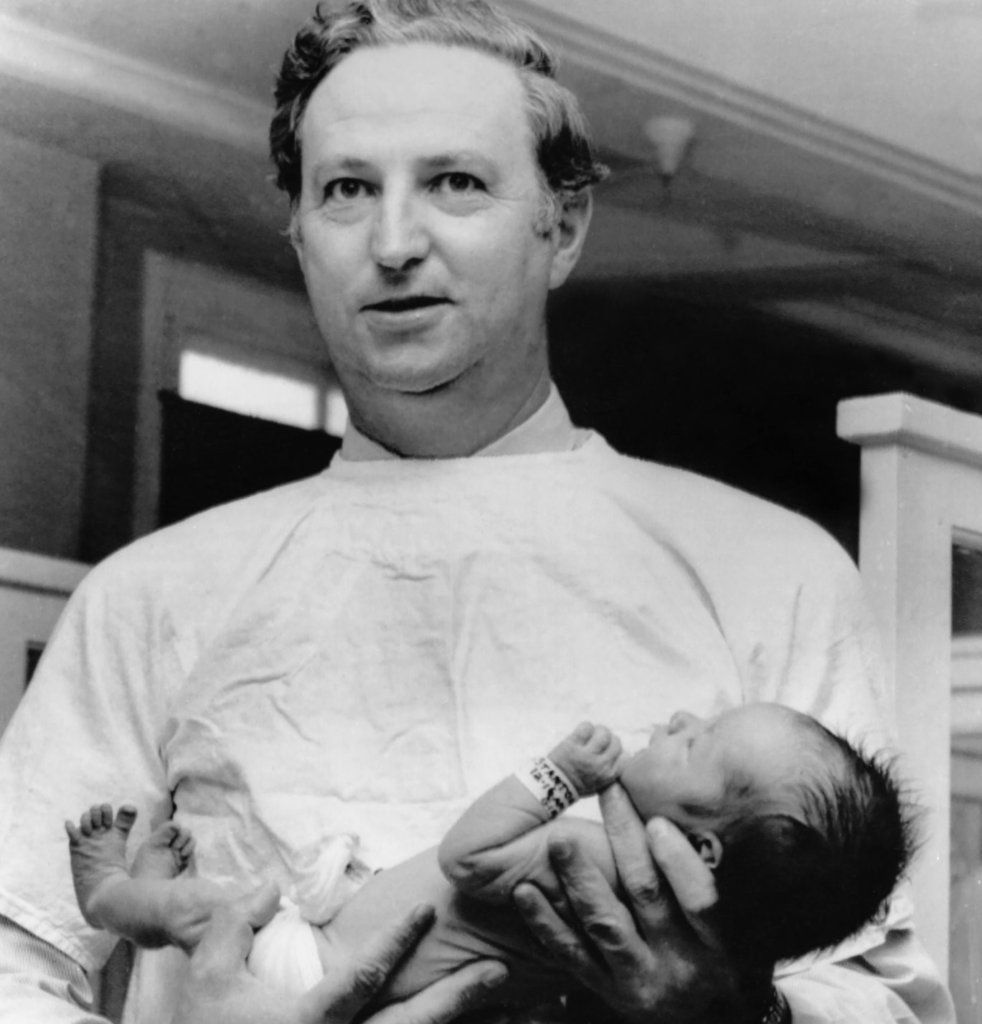
La Chemie Grunental, verso la fine del 1957 informò i medici della commercializzazione del suo sedativo, la talidomide.
La ditta combinò il farmaco con altri prodotti farmaceutici come l’aspirina, la fenacetina, il chinino ecc, tanto che i tedeschi assunsero talidomide per curare disturbi come raffreddore, tosse, influenza, nervosismo, nevralgie, emicrania ed asma.
Nel 1958 la campagna pubblicitaria del Contergan assunse proporzioni massicce: 50 inserzioni sulle riviste mediche, 200.000 lettere inviate ai medici; 50.000 “circolari terapeutiche” furono inviate ai medici e ai farmacisti.
Nella pubblicità della talidomide, commercializzata in più di 45 Paesi, si sottolineava la completa atossicità del farmaco.
Il 1 Agosto 1958 a 40.245 medici MG venne inviato uno studio del dottor Blasiu.
Nella lettera di accompagnamento la Chemie Grunental descriveva il Contergan come il miglior farmaco da somministrare alle gestanti e alle madri che allattano: farmaco che “non danneggia né la madre né il bambino”.
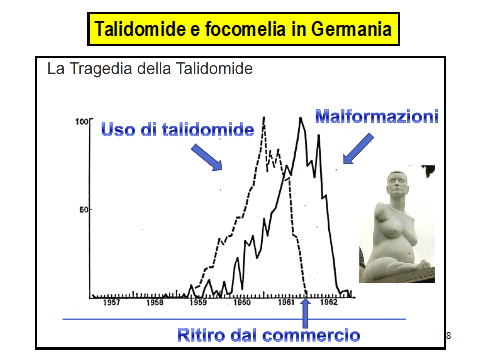
Nel 1961 la Chemie Grunental dovette affrontare la questione della sicurezza della talidomide usato durante la gravidanza.
Durante un congresso di pediatria nella Germania occidentale tenutosi a Dusseldorf il 18 novembre 1961 si parlò di una misteriosa esplosione di focomelia.
In seguito la casa farmaceutica ritira il farmaco dal mercato.

Digitando la parola talidomide nell’archivio del Corriere della Sera, dal 1960 a oggi, si trovano 489 articoli.
E’ curioso che uno, già nel 1961, parlasse di utilizzo del farmaco nella cura dei tumori.
Poi nel tempo ci sono stati articoli che hanno raccontato storie di persone nate con malformazioni
E molto si è scritto sulla questione degli indennizzi fino a oggi e fino a un ultimo articolo comparso su Sette a firma di Gian Antonio Stella: c’è tempo fino al 2018 per cercare le vittime.




Ma perché succede questo?
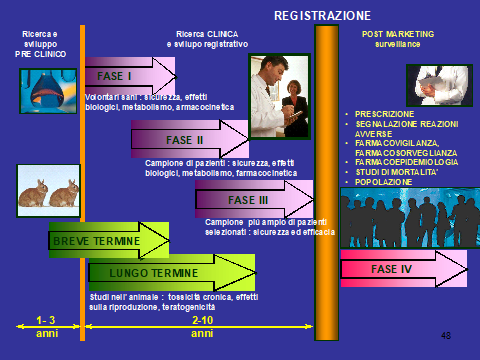


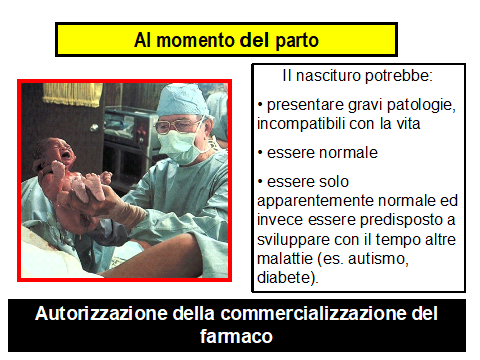
Con l’immissione in commercio di un nuovo farmaco avviene l’esposizione della popolazione generale, prima che ne siano del tutto noti gli effetti terapeutici e le reazioni avverse.
Gli studi di fase III, interessando popolazioni poco numerose, non sono in grado di evidenziare tutte le possibili reazioni avverse ai farmaci in studio, specie se rare.
Tali studi poi sono di durata troppo breve (3-12 mesi) per consentire di rilevare sia la tossicità cronica che quella ritardata.
Nell’uso corrente i farmaci sono impiegati su popolazioni più numerose e composte da pazienti che differiscono nettamente da quelli arruolati negli studi di pre -commercializzazione.
Deve essere valutato l’impatto dell’intervento farmacologico sul decorso naturale delle malattie e quindi la stima del livello di utilità del farmaco rispetto al costo per la collettività che ne fa uso.
Per chiarire i reali rapporti rischio/beneficio dei farmaci si è sviluppata una branca della farmacologia clinica definita farmacosorveglianza.
1. Introduzione
La Farmacovigilanza (FV) consiste nella valutazione del rischio e nel monitoraggio della incidenza di effetti indesiderati potenzialmente associati al trattamento farmacologico.
I quattro obiettivi principali della FV consistono nell’individuare il più celermente possibile nuove Adverse Drug Reactions (ADR), nel migliorare e rendere più adeguate le informazioni su ADR sospette o già note, nell’analizzare i benefici di un farmaco rispetto ad altre tipologie di terapia, nel trasmettere diffusamente tali informazioni per rendere più corretta e adeguata la pratica clinica terapeutica. Tali obiettivi appaiono di vasta portata, e, a ben guardare, coinvolgono molti degli ambiti della buona pratica clinica e molti dei criteri della Medicina Basata sulle Evidenze (Evidence Based Medicine – EBM).
In effetti essi riguardano la corretta definizione diagnostica alla luce delle migliori evidenze disponibili nella letteratura medica e dell’esperienza clinica individuale, l’adeguato provvedimento terapeutico in caso di ADR, le analisi di costo-beneficio e di costo-efficacia di un agente farmacologico, il confronto di tali rapporti con quelli di farmaci consimili, la creazione di banche dati contenenti le informazioni corrette e pertinenti, la disseminazione di tali informazioni al maggior numero di operatori al fine di migliorare la pratica clinica quotidiana1.
- Componenti e impatto sociale della Farmacovigilanza
Gli elementi che fanno della FV un campo particolarmente rilevante al giorno d’oggi non sono d’altronde solo quelli accennati poco sopra, ma altri fattori giocano un ruolo preminente nell’indicare il rilievo della FV per un numero sempre crescente di operatori sanitari e di utenti del sistema sanitario. In primo luogo il costante, continuo ed inarrestabile invecchiamento della popolazione generale. Dal 1960 al 1990 la popolazione complessiva degli Stati Uniti è aumentata del 39%, laddove il numero di soggetti al di sopra dei 64 anni è aumentato dell’89% e quello dei soggetti con più di 84 anni del 232%.
Nel 2010, il numero di cittadini canadesi con più di 64 anni è aumentato del 45% ed il numero degli ultra-ottantacinquenni del 125%.
Nella maggior parte dei Paesi industrializzati la quota di soggetti anziani cresce rapidamente, e parallelamente ad essa cresce il carico sociale di patologie croniche, curabili ma non guaribili, che necessitano di approcci farmacologici complessi, non di rado di poli-terapia. Il soggetto anziano dunque, soprattutto quando fragile, rappresenta il terreno su cui con maggiore probabilità si devono ricercare e attentamente monitorare reazioni indesiderate ai farmaci, un terreno che è reso ancora più complesso dall’invecchiamento fisiologico (se non patologico) dei meccanismi metabolici e detossificanti, dall’interazione non di rado imprevedibile di numerosi e concomitanti agenti farmacologici, dalle differenze di sesso e di razza.
L’impatto sociale delle ADR è quindi destinato a salire in termini numerici, ma anche in termini di gravità (in soggetti anziani fragili, come si accennava sopra) ed in termini di costi economici per la società. Il carico di morbilità delle ADR è notevole, dunque, se si pensa che numerose sono le analisi della letteratura e delle casistiche cliniche che indicano che, su grandi numeri di pazienti ricoverati in ospedale, una quota superiore al 5% viene ammessa proprio per ADR gravi.
Ancora più impressionante è il carico di mortalità, quando si leggono report aggiornati ed affidabili che segnalano come le ADR rappresentino addirittura la quarta-sesta causa di morte negli USA, superando in tale funesta graduatoria perfino il diabete mellito.
Tale quadro preoccupante è completato dalle proiezioni a breve termine, che prevedono un aumento generale del numero di casi e di decessi conseguenti ad ADR.
E’ molto probabile, peraltro, che i numeri attualmente disponibili siano sottostimati, dal momento che il sistema di diagnosi, monitoraggio, segnalazione e registrazione di effetti indesiderati a farmaci è ancora largamente migliorabile non solo in molti Paesi europei (in primis l’Italia), ma anche in molte aree degli Stati Uniti. In effetti il tema della sorveglianza intensiva e sistematica delle ARD è al giorno d’oggi al centro di un ampio dibattito, e le modalità di implementazione di tale sistema sono attualmente ampiamente discusse.
2. Leggi e norme2
Sulla Gazzetta Ufficiale n. 101 del 3 maggio 2003 è stato pubblicato il Decreto Legislativo dell’8 Aprile 2003, n. 95 “Attuazione della Direttiva 2000/38/Ce relativa alle Specialità Medicinali, Testo in vigore dal 18-5-2003”.
Art. 4….. “I medici e gli altri operatori sanitari sono tenuti a segnalare tutte le sospette reazioni avverse gravi o inattese di cui vengano a conoscenza nell’ambito della propria attività. Vanno comunque segnalate tutte le sospette reazioni avverse osservate, gravi, non gravi, attese ed inattese da tutti i vaccini e da farmaci posti sotto monitoraggio intensivo ed inclusi in elenchi pubblicati periodicamente dal Ministero della Salute. Il presente decreto non si applica alle segnalazioni di reazioni avverse verificatesi in corso di sperimentazione clinica.”Questo è uno dei punti più importanti del nuovo decreto legge, in quanto si viene a perdere l’obbligo di segnalazione per le reazioni non gravi e già conosciute.
Art. 4….. “I medici e gli altri operatori sanitari devono trasmettere le segnalazioni di sospette reazioni avverse, tramite l’apposita scheda, tempestivamente, al Responsabile di farmacovigilanza della struttura sanitaria di appartenenza.”
Per quanto riguarda le sanzioni stabilite nell’articolo 11 del decreto del 1997 il nuovo decreto non fa più riferimento a sanzioni per medici e farmacisti che non segnalino le sospette ADR. Al contrario, per il titolare dell’autorizzazione all’immissione in commercio, le sanzioni amministrative sono state elevate. Anche per i responsabili di farmacovigilanza delle strutture sanitarie che violino gli obblighi di segnalazione scompaiono le sanzioni penali, ma sono previste soltanto sanzioni disciplinari.
Nel complesso quindi, il nuovo decreto sembra voler porre l’attenzione alle reazioni avverse gravi ed inattese, dando maggior enfasi alla qualità (più che alla quantità) delle segnalazioni e quindi al ruolo della segnalazione spontanea come strumento per generare un segnale d’allarme.
Inoltre, l’abolizione delle sanzioni e dei limiti temporali imposti per le segnalazioni è intesa a valorizzare ulteriormente il ruolo della farmacovigilanza, rimuovendo quegli aspetti burocratici e vessatori che, tra l’altro, non hanno fornito alcun beneficio in termini di incremento del numero delle segnalazioni.
L’attuale decreto riprende la legislazione precedente apportandovi alcune modifiche. Le segnalazioni dei medici e dei farmacisti (questi ultimi per le reazioni avverse da farmaci non soggetti a prescrizione medica) devono essere inviate tempestivamente alle aziende sanitarie locali o alle direzioni sanitarie degli ospedali, le quali devono trasmetterle al Dipartimento per la valutazione dei medicinali e la farmacovigilanza del Ministero della sanità, informando anche la Regione di appartenenza e l’industria responsabile del medicinale. A sua volta il dipartimento deve poi informare l’Agenzia europea; non è previsto invece che i dati analitici ottenuti vengano riferiti alle ASL o alle Regioni, e quindi nemmeno tornino ai medici né ai farmacisti. Genericamente si demanda tale attività informativa alle Regioni, senza peraltro precisare se è prevista la possibilità di un accesso ai dati nazionali.
“….. la segnalazione spontanea delle reazioni avverse fa parte di un modo più attento di prescrivere i farmaci e rientra in un comportamento del medico che non si ottiene con le sanzioni, ma attraverso un’operazione di crescita culturale continua, seria e indipendente.
Pur sapendo che la realtà si modifica con la pratica piuttosto che con l’emanazione di norme, notevoli erano le aspettative per il nuovo decreto legislativo sulla farmacovigilanza. In particolare da più parti si auspicava un potenziamento delle attività decentrate e del flusso informativo di ritorno, prendendo a modello i sistemi nazionali di paesi, quali la Francia o il Regno Unito, che hanno ottenuto rilevanti successi in questo campo.”
Ogni medico è tenuto a segnalare sull’apposito modulo (DM 7 agosto 1997) tutte le sospette reazioni avverse gravi o inattese delle quali sia venuto a conoscenza nell’esercizio della propria attività. La normativa riguarda anche i farmacisti.
2 Farmacovigilanza e medicina basata sulle evidenze: un felice connubio metodologico
| Riferimenti legislativi |
| DM sanità 28.7.1984 |
| Legge 531/1987 |
| Direttiva europea 93-39 del 14.6.1993 art. 29 bis, art. 29 ter, art. 29 sexis |
| Regolamento europeo 2309/1993 |
| Legge 52/1966 |
| Decreto legislativo 44/1997 art. 2, art. 4, art. 11 |
| DM sanità 7.8.97 |
| Circolare MINSAN 15/1999 |
| Decreto Legislativo dell’8 Aprile 2003, n. 95 “Attuazione della Direttiva 2000/38/C |
La segnalazione va effettuata su apposito modulo nel quale obbligatoriamente devono essere riportate:
il nome commerciale del prodotto
la fonte della segnalazione(ospedale, medico di medicina generale, specialista, farmacista)
la reazione
la sua gravità
la data di inizio
l’età del malato
il sesso del malato
3. Come valutare la sicurezza di un farmaco
Per valutare la sicurezza di un farmaco è necessario mettere insieme tutte le informazioni raccolte da differenti fonti, quali:
- Studi pre-clinici;
- Trial clinici (pre e post–marketing);
- Segnalazioni spontanee di reazioni avverse (su base sia nazionale che internazionale);
- Studi epidemiologici (caso-controllo e di coorte);
- Dati raccolti per altri scopi (statistiche nazionali ed internazionali, banche dati di prescrizioni e di outcomes clinici).
Questo processo di valutazione inizia con gli studi di farmacologia preclinica e deve continuare fino a quando il farmaco esce dal mercato, periodo che può durare alcuni decenni. L’identificazione di un danno causato da un farmaco può avvenire sia durante il suo sviluppo che dopo la sua commercializzazione. In quest’ultimo caso, ci sono buone probabilità che vengano identificati nei primi mesi di introduzione nel mercato, come è successo nel caso del mibefradil. Talvolta, però, sono necessari anni per accumulare dati sufficienti ad affermare che un farmaco è pericoloso, come è stato nel caso della fentermina.
Le principali ragioni per le quali il danno causato da un farmaco (reazione avversa da farmaco, ADR) viene individuato solo nel periodo post-marketing sono le seguenti:
- La reazione avversa è rara e quindi non identificabile prima che un gran numero di pazienti venga esposto al farmaco.
- Esiste una lunga latenza fra la somministrazione del farmaco e lo sviluppo della reazione avversa (es. ipertensione polmonare da fentermina).
- Il farmaco non è stato studiato nella normale pratica clinica. Infatti i pazienti trattati nella normale pratica clinica hanno buone probabilità di avere caratteristiche differenti (es. altre malattie, farmaci concomitanti, ecc.) da quelli dei trials clinici, dove tra l’altro i farmaci vengono usati in accordo con le raccomandazioni dei medici, con il consenso dei pazienti e sotto stretto controllo.
4. Il segnale
E’ necessario in primo luogo che venga identificato un evento indesiderato (danno) e che questo sia dipendente dal farmaco. E’ evidente che nelle fasi iniziali non si può essere certi che danno e farmaco siano correlati.. Questa incertezza ha portato a sviluppare il concetto di “segnale”, che è definibile come una richiesta di “attenzione” lanciata da qualsiasi fonte (esempio un case report), che ha sospettato che un farmaco possa essere associato con un rischio precedentemente ignoto o che un rischio già noto possa essere quantitativamente o qualitativamente differente da quello che ci si deve aspettare. Termini alternativi presenti in letteratura ed utilizzati come sinonimi sono “allarme” o “generazione di una ipotesi”.
5. La segnalazione spontanea
La segnalazione spontanea di una ADR può pertanto essere considerata come un classico sistema di segnale ed il suo scopo principale è quello di fornire il più presto possibile l’allarme di un possibile rischio. In altri termini la segnalazione spontanea di una ADR è la descrizione di un evento clinico non previsto e/o non desiderato che colui che effettua la segnalazione ritiene che possa essere collegato al farmaco o ad i farmaci che vengono assunti.
Il sistema della segnalazione spontanea non chiede ai segnalatori di riportare tutti gli eventi avversi che seguono alla somministrazione dei farmaci, ma di riportare selettivamente solo quelli che si sospetta siano causati dal farmaco.
Le ragioni per cui un segnalatore può sospettare che un farmaco possa aver causato la reazione avversa possono essere diverse, fra cui una o più delle seguenti:
- Associazione temporale. Esiste un intervallo di tempo plausibile fra l’assunzione del farmaco e l’insorgenza del possibile evento avverso.
- Dechallange. La sospensione del farmaco entro un lasso di tempo plausibile ha portato alla riduzione o alla scomparsa dell’evento avverso.
- Dose-risposta. L’aumento della dose o la sua riduzione ha provocato un aumento della severità o una riduzione o la scomparsa dell’evento.
- Rechallange. L’eventuale nuova somministrazione del farmaco ha portato alla comparsa dello stesso evento avverso.
- Meccanismo d’azione. Il meccanismo d’azione del farmaco può rendere ragione della comparsa dell’evento avverso.
- Effetto di classe. L’evento avverso che si ritiene imputabile a quel farmaco è già stato descritto per farmaci della stessa classe (con lo stesso meccanismo d’azione).
- Assenza di alternative. L’evento avverso non è spiegabile in base allo stato di malattia del paziente o in base ad altri farmaci assunti nel presente o nel passato.
Nessuno dei fattori sopra menzionati, di per sé, è essenziale per portare a sospettare una relazione di causalità, ma più ce ne sono insieme maggiore è la probabilità. Tuttavia un sospetto molto forte non porta di per sé ad una maggiore probabilità di segnalazione poiché la conoscenza di un meccanismo d’azione e degli effetti avversi di farmaci simili può far sì che il segnalatore dia per scontato l’evento e non lo segnali.
6. La sottosegnalazione
La sottosegnalazione o “under-reporting” è il fenomeno più comune di tutti i sistemi di segnalazione spontanea.
Un sistema efficace di segnalazione spontanea deve prevedere tre aspetti:
- Facilitazione della segnalazione e riscontro di ciascuna segnalazione al segnalatore (feedback).
- Educazione continua sulla sicurezza dei farmaci e sui benefici che il sistema di segnalazione spontanea può apportare alla comunità sia in termini di riduzione del rischio per il paziente che di risparmio delle risorse per curare le reazioni avverse.
- Progressivo coinvolgimento dei segnalatori in una rete permanente di farmacovigilanza con rapida messa a disposizione dei risultati del sistema.
7. Evento avverso e reazione avversa.
Esiste una confusione fra le definizione di “evento avverso” e di “reazione avversa” .
La prima definizione dovrebbe essere usata nel contesto di studi dove tutti gli eventi vengono raccolti indipendentemente dal fatto che siano o meno sospettabili come collegati ad un farmaco.
La seconda dovrebbe essere sempre accompagnata dall’aggettivo “sospetta” quando si riferisce ad un caso o ad una serie di casi che vengono raccolti attraverso un sistema di segnalazione spontanea.
Questo approccio è in accordo alle definizioni date dalla Comunità Europea (Chapter Va Council Directive 75/319/EEC, come corretto dal 93/39) ed alla proposta della International Conference on Harmonisation (ICH) nella linea guida E2A(2).
8. Il segnale d’allarme.
Come già detto, nella fase iniziale di un processo di valutazione della sicurezza di un farmaco non si può essere certi che il danno sia dipendente dal farmaco.
Questa incertezza ha portato a sviluppare il concetto di “segnale”. Tuttavia per generare un “allarme” non basta una sola segnalazione (3). Bisogna in primo luogo verificare se esista o meno un “inaspettato” numero di eventi avversi correlati al farmaco.
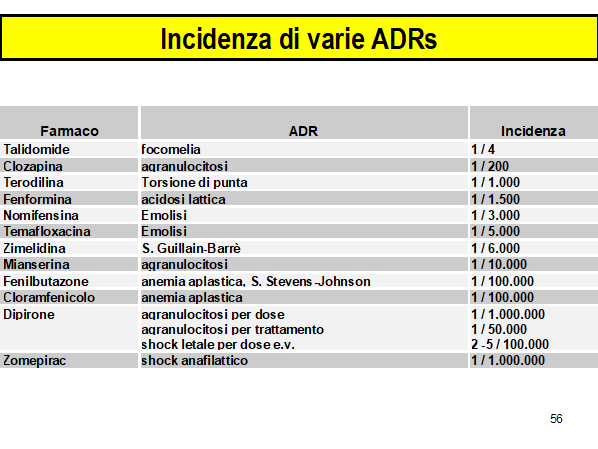
Quando l’ADR è un evento raro nella popolazione, come ad esempio la necrolisi epidermica tossica o l’anemia aplastica, un piccolo numero di casi associati con un singolo farmaco è improbabile che rappresenti un evento causale, anche se il farmaco è molto usato (4). In questa condizione tre casi possono essere considerati un segnale di allarme e cinque casi un segnale forte di allarme. Una spiegazione di questo concetto è riportata nella Tabella I.
Se l’evento è frequente nella popolazione generale, saranno necessarie molte segnalazioni per sollevare un allarme (5) e l’esempio della Tabella I non è applicabile. Un giudizio sul pericolo potrà essere fatto ricorrendo a tutte le altre informazioni disponibili, fra cui la forza della evidenza.
Sono disponibili numerosi metodi da applicare per determinare se le segnalazioni spontanee può generare un allarme.
- Il consumo del farmaco. Si può porre come denominatore il consumo del farmaco sospettato, sia come numero di prescrizioni che come dosi definite giornaliere (DDD) ed in tal modo valutare con quale probabile frequenza l’evento può manifestarsi. Tuttavia a causa del variabile ed indeterminabile grado di under-reporting tali valutazioni debbono essere molto accurate. Infatti potremmo trovarci a confrontare farmaci messi in commercio con indicazioni differenti o per periodi di tempo diversi o per i quali vi è stata una differente informazione (o pubblicità) sugli eventi avversi possibili.
- Il profilo delle reazioni avverse (1). Si calcola per un dato farmaco la proporzione di reazioni che ha interessato un particolare organo od apparato (Tabella II). Questo metodo ha il vantaggio di essere indipendente dal consumo del farmaco.
- Rapporti proporzionali di segnalazioni o PRRs (6). Si utilizzano i calcoli di tabelle 2 x 2 che confrontano la proporzione di tutte le ADRs del farmaco che ci interessa (es. uveite da rifabutina), presenti in un determinato database, con l’analoga proporzione della stessa ADR indotta da tutti gli altri farmaci (Tabella III). La significatività viene calcolato con il test χ2 e possono essere stabiliti dei criteri per l’automatica generazione dell’allarme (es. PRR > 3; χ2 > 5, almeno tre segnalazioni). La natura matematica di questo approccio non dovrebbe essere usata per ridurre l’intrinseca natura dei dati e le limitazioni date dalla possibile casualità.
9.La qualità della segnalazione
Ci sono quattro argomenti che rendono un segnale qualitativamente importante e che determineranno se verrà o meno ulteriormente studiato:
- La forza della segnalazione. Una segnalazione accurata in cui il caso clinico è ben descritto (eventualmente corredato da dati strumentali e di laboratorio ed alla presenza o assenza di altre patologie), in cui l’associazione temporale fra esposizione al farmaco (con dose, via di somministrazione, durata, altri farmaci associati) è accuratamente riportata, in cui è descritto l’effetto del dechallenge (e dell’eventuale rechallenge) ha molto più valore di una segnalazione incompleta.
- La novità. L’evento correlato al farmaco non è mai stato descritto o non è presente in letteratura; la segnalazione riguarda un farmaco entrato di recente in commercio; si riferisce a popolazioni poco o per nulla studiate nel trials clinici pre marketing (donne gravide, bambini, anziani); si sospetta che la reazione avversa sia probabilmente correlata a terapie non accettate ancora dalla comunità scientifica occidentale (fitoterapia, agopuntura, ecc.),
- L’importanza clinica.Viene giudicata in base alla gravità dell’evento osservato ed in base alla sua rarità.
- La potenzialità. Rappresenta la potenzialità che tale segnale ha di determinare l’assunzione di misure preventive. Ad esempio, è possibile che l’evento avverso sia correlato ad una interazione tra il farmaco sospettato e un altro farmaco che il paziente assume.
10. Provenienza del segnale.
Il segnale può derivare da segnalazioni spontanee o da studi formali e l’importanza di esso varierà in maniera sostanziale.
Segnale proveniente da segnalazioni spontanee.
In molti casi il segnale all’inizio consisterà di una serie di segnalazioni di eventi avversi che sono insorti a seguito della assunzione di un particolare farmaco. In questo caso dovranno essere presi in considerazione una serie di fattori per valutare il significato di tale segnale. Bisogna valutare singolarmente tutte le segnalazioni (Tabella IV) e cercare altre evidenze (Tabella V).
- La valutazione della causalità può essere fatta con vari metodi (7) e molto usati sono i vari algoritmi disponibili. Tuttavia è indispensabile escludere che esistano spiegazioni alternative che giustifichino l’evento (Tabella VI). In poche parole si tratta di effettuare una accurata diagnosi medica differenziale. Semplificando, bisogna chiedersi se l’evento avverso trova spiegazione nel farmaco usato o una malattia concomitante possa aver determinato detto evento.
- Possibili alternative farmaco-correlate.
- paziente esposto a due farmaci, ciascuno dei quali noto come capace di indurre l’evento indesiderato in questione. Il segnalatore può sospettare uno o l’altro dei due farmaci o entrambi, ma è probabile che riporti l’evento come dovuto al farmaco di più recente commercializzazione. Chiaramente è possibile che l’evento sospettato ed imputato al farmaco nuovo non sia dovuto a quest’ultimo, ma potrebbero esserci diverse ragioni per le quali viene sospettato il nuovo farmaco. In particolare il segnalatore potrebbe non sapere che l’ADR segnalata non è nuova o potrebbe avere alcune buoni ragioni per sospettare il nuovo farmaco, in base alla relazione temporale fra assunzione del farmaco e comparsa della reazione.
- paziente esposto a due farmaci, nessuno dei quali noto come capace di indurre l’evento indesiderato in questione. In questo caso può essere sospettato uno solo dei due, spesso poiché l’evento avverso insorge entro breve tempo dalla sua introduzione o perché il farmaco è di recente commercializzazione. In questa circostanza è possibile formarsi una opinione della causa più probabile o della combinazione di cause, basandosi sulla temporalità dell’evento e sul dechallange. Spesso, tuttavia, non è possibile dare un giudizio definitivo.
- paziente esposto ad una interazione tra farmaci. Le possibili interazioni tra farmaci sono difficili da interpretare quando provengono dalle segnalazioni spontanee. Di norma il numero di segnalazioni è basso, ma il sospetto può essere rafforzato se si possono valutare i seguenti punti: 1) esistenza o meno di documentazione scientifica di interazioni per il farmaco sospettato; 2) presenza nella segnalazione di dati di laboratorio, che forniscano misure di cinetica del farmaco sospettato, sia quando somministrato da solo che quando somministrato in congiunzione con altri farmaci; 3) sulla base delle conoscenze farmacocinetiche e farmacodinamiche dei farmaci assunti è ipotizzabile un meccanismo che non è stato studiato nei test preclinici di interazione (in questo caso tali studi dovrebbero essere immediatamente condotti); 4) il farmaco sospettato ha un basso indice terapeutico e l’ADR è grave.
- Possibili alternative non correlate ai farmaci.
Una o più delle sospettate ADR segnalate potrebbero non essere eventi dovuti al farmaco, ma una naturale conseguenza della malattia, correlabile o meno alle indicazioni per cui il farmaco “falsamente” sospettato è indicato. Poiché la maggior parte delle ADR sono simili alle malattie naturali, è evidente che bisogna tenere presente alcuni particolari:
- rarità nella popolazione dell’evento osservato. Una serie di casi di eventi rari che insorgano in relazione all’esposizione ad un particolare farmaco rappresentano una evidenza di causalità molto più forte di una serie di casi di eventi frequenti nella popolazione. Ciò non significa che il farmaco ha minori probabilità di essere la causa dell’evento, ma che tale rapporto di causalità è molto più difficile da stabilire.
- evento avverso come complicazione della malattia. Quando l’ADR sospettata è una complicazione della malattia per cui il farmaco viene usato (vedi esempio in Tabella VI), la segnalazione spontanea ha scarsa possibilità di risultare utile. Non è possibile con la segnalazione spontanea mettere in evidenza che un farmaco possa aumentare il rischio di complicazioni (es. aritmia da riperfusione dopo streptochinasi). Sono necessari studi formali che misurino tale rischio.
- Un gran numero di segnalazioni, molto poco documentate, avranno scarso valore. Tuttavia se l’evento riportato da tutte queste segnalazioni fosse grave, un’ulteriore valutazione sarebbe opportuna. Inoltre, quanto maggiore è il numero dei casi in relazione all’uso e quanto più completamente essi sono documentati, tanto più sarà necessario studiare il problema di sicurezza di questo farmaco.
In conclusione, se la segnalazione spontanea fornisce una serie di casi ben documentati di una particolare sospetta ADR senza ovvie spiegazioni alternative e/o con l’evidenza di un possibile meccanismo, è necessario prendere rapidamente in considerazione : 1) quali studi formali iniziare onde cercare una conferma dell’allarme; 2) quali azioni legislative o terapeutiche mettere in atto per minimizzare il rischio per la popolazione.
Al contrario, se la segnalazione spontanea fornisce casi clinicamente poco seri e già noti, per di più mal documentati e nella maggior parte dei casi con spiegazioni alternative, allora essa cessa la sua funzione di scatenare un allarme precoce.
Segnale proveniente da studi formali.
L’allarme che scaturisce dagli studi formali è molto più forte di quelli derivati dalla segnalazione spontanea. Infatti il disegno dello studio formale e l’analisi dei dati può portare ad una valutazione molto migliore della causalità e della frequenza dell’evento avverso. Tuttavia, nella fase iniziale della valutazione di un allarme è spesso presente un solo studio. Questo, anche se accuratamente disegnato e con dati apparentemente chiari, quasi mai permette di dare risposte sufficienti (8,9).
Il fattore che stimola a condurre uno studio formale è quasi sempre un allarme rappresentato dalla maggiore frequenza con cui l’evento avverso compare in presenza di un farmaco, rispetto ad un altro farmaco di confronto o rispetto alla normale incidenza di tale evento nella popolazione.
Lo studio che valuta l’aumentato rischio deve essere in grado di chiarire se esista o meno una correlazione fra assunzione del farmaco ed evento avverso. Per valutare la causalità però bisogna escludere i fattori principali che possono portare a falsi positivi. Essi sono:
- Casualità. Per dimostrare che l’evento avverso non è casuale è necessario che lo studio abbia il potere di condurre ad una differenza statisticamente significativa. Bisogna anche sapere se, a priori, è stata fatta o meno una ipotesi.
- Bias. Esistono molti tipi di bias (distorsione sistematica dei risultati) che possono influenzare i risultati di studi formali, siano essi trials clinici o studi epidemiologici. E’ importante pertanto valutare attentamente come sono stati reclutati i pazienti posti in trattamento e come sono stati raccolti i dati.
- Effetti di confusione. Bisogna conoscere quali fattori (es. fumo, alcool, patologie concomitanti, ecc) possono spiegare le differenze fra i gruppi, ad di fuori del trattamento farmacologico. Bisogna stabilire anche i provvedimenti da prendere per controllare questi fattori di confusione nel disegno dello studio e nell’analisi dei dati. Quando i dati vengono corretti per i fattori di confusione è importante valutare sia il rischio non corretto, che quello corretto e quest’ultimo deve essere tenuto in maggior considerazione del primo.
- Causalità. Una volta esclusi casualità, bias e fattori di confusione come spiegazioni alternative dell’evento, è possibile stabilire il nesso di causalità con il farmaco. Meglio ancora se in questo processo di valutazione si può far ricorso ad evidenze da altre informazioni (es. meccanismo d’azione) che possono rinforzare o spiegare questo rapporto. Sono stati descritti dei criteri formali per la valutazione della causalità (10), ma al momento sembra che una metanalisi, che prenda in considerazione le evidenze sia di trial clinici che di studi epidemiologici (11), sia il meglio disponibile per effettuare tali valutazioni.
Segnale proveniente da studi epidemiologi.
Per condurre uno studio epidemiologico atto a verificare la sicurezza di un farmaco sono necessarie delle banche dati che contengano a) dati di esposizione al farmaco; b) dati sugli effetti del farmaco, nonché la possibilità di accedere a dettagliate informazioni mediche di tutti i singoli casi.
Banche dati
I dati di esposizione e di effetti del farmaco possono essere contenuti nello stesso database, come nel caso del General Practice Research Database inglese, o in database separati, che possono essere collegati, come nel caso DSRU inglese (Tayside Prescription-Event Monitoring). L’elemento chiave di tale collegamento, detto record-linkage, è l’identificazione crociata degli individui nei due database, in modo tale che le esposizioni al farmaco e gli effetti dello stesso siano collegabili al singolo paziente.
Va tenuto presente che nessuna fonte di dati è perfetta, per cui la qualità e l’accuratezza del dato sono importanti considerazioni da fare quando si stabilisce di condurre una indagine. La mancanza di accuratezza del dato non impedisce di trovare una correlazione tra evento e causa, ma può produrre una tale diluizione della associazione evento-causa da portare a risultati incerti. L’inaccuratezza del dato porterà ad una apparente associazione positiva, solo se sono presenti bias grossolani e sistematici. Un modesto grado di inaccuratezza nei dati ha poche probabilità di produrre una falsa positività se l’associazione tra evento e causa esiste.
Il database di esposizione al farmaco negli studi epidemiologici è sempre imperfetto (14). Infatti in nessun caso vi sarà la certezza che il farmaco prescritto al paziente sarà stato da questo realmente assunto nelle modalità descritte. Tale problema può essere valutato intervistando direttamente i pazienti, ma anche in questo caso non è detto che i pazienti descrivano correttamente il loro comportamento.
Il database sugli effetti del farmaco è anch’esso sempre carente in qualità ed accuratezza (14). Non è un caso infatti che è necessario sempre verificare la cartella clinica per pulire i dati, eliminando lentamente gli errori fatti durante la fase di inserimento nel database. Tuttavia, la stessa pulitura del dato crea un grande svantaggio che è rappresentato dal fatto che spesso vengono eliminati o non caricati i dati non ben documentati. Si determina in tal modo un bias di selezione e l’incidenza dell’evento probabilmente risulterà sottostimata.
Un particolare problema connesso al disegno ed alla interpretazione di molti studi epidemiologici sulla sicurezza dei farmaci è quello relativo all’utilizzo selettivo o meno del farmaco, oggetto della ricerca, in pazienti che hanno caratteristiche diverse da quelli che vengono usati come controlli. E’ ormai noto che questo è un problema comune ed in parte connesso alle strategie messe in atto dal marketing delle aziende farmaceutiche. Per esempio, far credere che un farmaco antiinfiammatorio non steroideo abbia minor tossicità gastrointestinale di un altro della stessa classe, può far sì che questo venga usato selettivamente in pazienti con una storia pregressa di tossicità gastroenterica. Uno studio epidemiologico su un database, che contenga questi casi, porterà con una certa probabilità a dimostrare che questo farmaco è associato paradossalmente ad un maggior rischio di tossicità gastroenterica dei FANS di confronto (5).
Nonostante tutte le limitazioni sopra riportate, i bassi costi e la velocità con cui si possono ottenere i risultati ha portato, negli ultimi anni, all’utilizzo sempre maggiore delle banche dati disponibili al fine di valutare la tossicità dei farmaci. Queste banche dati, che non sono state create per raccogliere dati sulla tossicità dei farmaci, vengono oggi utilizzate per condurre due tipi principali di studi epidemiologici: di coorte e caso-controllo.
- Studi di coorte.
In questo tipo di studi, una ben definita popolazione viene seguita nel tempo e viene misurato l’incidenza dell’evento avverso in questione, confrontandolo negli individui esposti ed in quelli non esposti al farmaco in questione. Usando questo approccio, si può quantificare il rischio assoluto ed il rischio relativo.
- Studi caso-controllo.
In questo tipo di studi vengono identificati i casi con l’evento avverso di interesse e confrontati con un gruppo di controllo selezionato dalla stessa popolazione che non ha presentato quegli eventi. Sia nei casi che presentano l’evento che nei controlli si confronta l’eventuale precedente esposizione al farmaco. Questi studi forniscono misure di rischio relativo sotto forma di odd ratios, ma non misurano il rischio assoluto.
Un ulteriore approccio è quello di collegare uno studio caso-controllo ad un disegno di coorte. Questo approccio ha il vantaggio di entrambi i metodi, particolarmente se viene usato per facilitare un più dettagliato controllo sui potenziali fattori di confusione che possono essere presenti in una coorte completa.
11. Conseguenze dell’accertamento del rischio
La natura dell’azione da intraprendere in relazione alla verificata esistenza di un rischio dipenderanno da:
- Gravità dell’ADR.
- Frequenza dell’ADR
- Evitabilità della ADR
- Natura della malattia che viene trattata con il farmaco responsabile
- Benefici ottenibili con il farmaco responsabile dell’evento avverso
- Disponibilità o meno di terapie alternative.
In base a questi fattori l’azione da prendere potrà andare dal ritiro del farmaco dal mercato alla semplice diffusione dell’informazione di tale rischio agli operatori sanitari ed ai pazienti.
Inoltre, se il farmaco rimane in commercio, andranno valutate una serie di azioni (Tabella VII) che possono eventualmente prevenire questo evento avverso e renderne più sicuro l’uso nella popolazione.
12. Classificazioni delle reazioni avverse da farmaci
Le ADR possono essere classificate in reazioni tipo A, attese, reazioni tipo B o bizzarre, imprevedibili ed inattese.
Reazioni tipo A : sono dose-dipendenti e correlate alla normale attività farmacodinamica di una molecola. Possono verificarsi in qualsiasi individuo. L’effetto farmacologico che provoca la reazione può coincidere con l’azione terapeutica ricercata, sebbene quantitativamente abnorme.
L’insulina, ad es., controlla il diabete mellito ma può scatenare una crisi ipoglicemica. Gli alchilanti, legandosi al DNA, esercitano la loro azione antiproliferativa ma, tramite lo stesso meccanismo, possono condizionare la comparsa di seconde neoplasie anche a distanza di anni.
Le reazioni di tipo A sono facilmente prevedibili e di solito riportate nelle ricerche farmacologiche e tossicologiche e/o negli studi clinici controllati. Le reazioni di tipo A sono evitabili o almeno minimizzabili nelle loro conseguenza previo adattamento delle dosi alle condizioni fisiologiche e/o patologiche dei singoli pazienti (età, stati di malattia, efficienza metabolica, funzionalità degli emuntori renale ed epatico) che potrebbero contribuire al verificarsi del fenomeno dell’iperdosaggio relativo. Una misura della capacità di produrre reazioni dose-dipendenti è data dall’indice terapeutico (intervallo fra dose terapeutica e dose tossica) dei singoli farmaci. Nel computo dell’indice terapeutico sono escluse le reazioni di tipo B. Le reazioni di tipo A sono molto frequenti al punto da costituire un importante fenomeno di salute pubblica dato che si è stimato che possano contribuire al 3-15% dei ricoveri ospedalieri e che si verificano nel 6-20% dei pazienti ospedalizzati. Tuttavia dal punto di vista clinico non sono di solito gravi, migliorano riducendo la dose di farmaco e sono solo eccezionalmente mortali. Sono di solito prevedibili con gli studi tossicologici sugli animali.
Reazioni di tipo B : non possono essere spiegate sulla base delle caratteristiche del farmaco che le determina perché derivano da una risposta ai farmaci inattesa, insolita, nuova e aberrante, correlata strettamente al terreno particolare del paziente nel quale si verificano. Sono più rare e non dipendono dalla dose somministrata. Clinicamente sono quasi sempre inevitabili, gravi e associate ad elevata mortalità. La loro insorgenza richiede trattamenti specifici e la sospensione del farmaco. Il paziente deve essere informato del fatto che una nuova somministrazione potrà scatenare reazioni del tutto sovrapponibili a quelle già sperimentate o ancora più gravi. Nella maggior parte dei casi la patogenesi è attribuibile a fenomeni di idiosincrasia o di allergia, ma i meccanismi sono meno conosciuti rispetto a quelli delle reazioni tipo A. Uno dei motivi principali è rappresentato dal fatto che questi effetti non vengono evidenziati dai test tossicologici preclinici sugli animali, sono difficili da riprodurre sperimentalmente in laboratorio e non sono ancora stati sviluppati soddisfacenti test predittivi.
| CARATTERISTICHE | Tipo A | Tipo B |
Effetto farmacologico | Aumentato | Bizzarro |
| Predittività | Si | No |
| Dose-dipendenza | Si | No |
| Morbilità | Elevata | Bassa |
| Mortalità | Bassa | Elevata |
ESEMPI DI ADR DI TIPO A E DI TIPO B
| Farmaci | tipo A | tipo B |
Antidepressivi | Sedazione, effetti anticolinergici | Epatotossicità |
| Ansiolitici/ipnotici | Sedazione, sintomi da sospensione | Epatotossicità |
| Levodopa | Nausea, vomito, discinesia, psicosi | Anemia emolitica |
| Beta-bloccanti | Bradicardia, ipotensione, broncospasmo | S. oculomucocutanea |
| Tiazidici | Ipocaliemia, gotta | Piastrinopenia, vasculite |
| Penicillina | Diarrea, convulsioni | Anafilassi, rush |
| Tetracicline | Uremia, discromie dentali | Fotosensibilizzazione, S. di Fanconi |
| FANS | Erosioni gastriche | Aplasia midollare |
Reazioni di tipo C
I farmaci, specialmente quando assunti per periodi di tempo molto prolungati (alcuni anni o per il resto della vita), possono indurre nuove malattie o modificare l’incidenza di una malattia.
Esempi possono essere identificate con la possibile incidenza di tumori del seno indotta dai contraccettivi orali o con la relativa frequenza di complicazioni tromboemboliche con differenti contraccettivi orali.
Questi eventi possono essere seri e relativamente comuni e possono influenzare significativamente la salute della popolazione.
L’insorgenza tardiva della malattia può rendere difficile individuarla come una patologia farmaco-correlata.
La relazione temporale spesso non è di nessun aiuto, così che può risultare impossibile provare o respingere il nesso di causalità.
Se poi il farmaco è ampiamente usato ed il numero assoluto di pazienti affetti dalla patologia sospettata come reazione avversa è elevato, la frazione aggiuntiva di incidenza di malattia indotta dal farmaco potrebbe essere troppo piccola per permettere di provare (o respingere) il nesso di causalità.
La valutazione a lungo termine dei benefici e dei danni causati dal trattamento delle malattie croniche richiede tempo e creazione di banche dati di morbidità e farmacoutilizzazione (Health Search).
13. VIGILANZA SUI FARMACI DA BANCO
La denominazione di “farmaci da banco” comprende un insieme di prodotti medicinali che possono essere ottenuti in farmacia senza prescrizione medica e utilizzati per l’automedicazione.
Nelle società ad elevato indice di sviluppo culturale e tecnologico il ricorso all’automedicazione è un fenomeno in continua espansione.
Le ragioni alla base dell’autogestione delle terapie mediche sono principalmente due: 1) l’automedicazione viene consentita per patologie minori, che incidono per brevi periodi di tempo sullo stato di salute e non sono caratterizzate da una prognosi sfavorevole; 2) si ritiene che i farmaci da banco utilizzati per l’automedicazione siano dotati di un buon profilo di efficacia e che, dal punto di vista della sicurezza d’impiego, essi siano relativamente “innocui”.
Leggi e norme1
La normativa europea in materia di farmacovigilanza è stata modificata con l’adozione nel 2010, del Regolamento UE 1235/2010, la cui applicazione è operativa dal 2 luglio 2012, e della Direttiva 2010/84/UE, attualmente in fase di recepimento.
E’ stato stimato che il 5% di tutti gli accessi in ospedale sono dovuti a reazioni avverse (ADRs), che il 5% di tutti i pazienti già ricoverati in ospedale presenta una ADR, che le ADR sono al quinto posto tra le cause di morte in ospedale. Pertanto, si è reso necessario intervenire sulle normative in vigore al fine di promuovere e proteggere la salute pubblica riducendo il numero e la gravità delle ADR e migliorando l’uso dei medicinali attraverso diversi tipi di intervento.
Fondamentalmente, i cambiamenti introdotti tendono ad aumentare l’efficacia, la rapidità e la trasparenza degli interventi di farmacovigilanza attraverso regole che mirano a:
- rafforzare i sistemi di farmacovigilanza, (ruoli e responsabilità chiaramente definiti per tutte le parti)
- razionalizzare le attività tra gli Stati Membri ad esempio attraverso una ripartizione delle stesse attività con condivisione del lavoro svolto evitando duplicazioni
- incrementare la partecipazione dei pazienti e degli operatori sanitari
- migliorare i sistemi di comunicazione delle decisioni prese e darne adeguata motivazione
- aumentare la trasparenza
Data la complessità delle attività da svolgere e dei cambiamenti da effettuare le modifiche saranno rese esecutive in tempi successivi, iniziando dai cambiamenti a maggior impatto sulla tutela della salute pubblica.
In primo luogo, cambia la definizione di reazione avversa intesa ora come “Effetto nocivo e non voluto conseguente all’uso di un medicinale”. Di fatto, con tale definizione, che è indipendente dal tipo di uso del medicinale, saranno oggetto di segnalazione le reazioni avverse, incluse anche quelle derivanti da errore terapeutico, abuso, misuso, uso off label, sovradosaggio ed esposizione professionale. Pertanto si avrà un incremento delle segnalazioni a cui corrisponderà una maggiore attività di monitoraggio.
In tutti i paesi dell’UE i pazienti potranno segnalare direttamente le sospette reazioni avverse. In Italia questa possibilità è già prevista da anni mediante modulo cartaceo, ma d’ora in avanti, in accordo anche alla nuova direttiva, le segnalazioni potranno essere effettuate anche via web.
Tutte le segnalazioni di reazioni avverse confluiranno nel database europeo Eudravigilance ma con una tempistica diversificata a seconda della gravità della reazione (entro 15 giorni per le segnalazioni gravi ed entro 90 giorni per quelle non gravi) e saranno accessibili al pubblico. Nel database Eudravigilance confluiranno anche le segnalazioni da parte delle aziende farmaceutiche. I dati delle reazioni avverse saranno resi accessibili e per alcuni medicinali autorizzati all’immissione in commercio con procedura centralizzata europea è già possibile consultare il database europeo delle reazioni avverse.
Il monitoraggio dei dati raccolti nel database Eudravigilance sarà effettuato dall’Agenzia Europea dei Medicinali in cooperazione con gli Stati Membri, mentre il monitoraggio dei dati originati a livello nazionale sarà effettuato dallo Stato Membro coinvolto; queste attività sono finalizzate all’identificazione di cambiamenti di rischi o di nuovi rischi attraverso l’analisi dei segnali, intendendo con questo termine “un’informazione proveniente da una o più fonti, osservazioni ed esperimenti compresi, che lascia supporre l’esistenza di una nuova associazione potenzialmente causale, o di un nuovo aspetto di un’associazione nota, tra un intervento e un evento o una serie di eventi collegati, avversi o benefici, ritenuta sufficientemente probabile da giustificare una verifica”. La metodologia per l’identificazione ed il processo di gestione del segnale sono stati definiti nel Regolamento di Esecuzione (UE) 520/2012 del 19 giugno 2012 relativo allo svolgimento delle attività di farmacovigilanza previste dal Regolamento (CE)n.726/2004 del Parlamento europeo del Consiglio e della Direttiva 2001/83/CE del Parlamento europeo e del Consiglio.
La nuova normativa è improntata anche ad una maggiore trasparenza e a migliorare la comunicazione. E’ stato previsto infatti che siano resi disponibili al pubblico, attraverso il portale web:
- rapporti di valutazione pubblici, unitamente a una loro sintesi
- riassunti delle caratteristiche del prodotto e fogli illustrativi
- riassunti dei piani di gestione del rischio
- elenco dei medicinali sottoposti a monitoraggio addizionale
- informazioni sulle diverse modalità per la segnalazione di sospette reazioni avverse dei medicinali alle autorità competenti da parte degli operatori sanitari e dei pazienti, compresi i moduli con maschera web di inserimento dati
I medicinali sottoposti a monitoraggio addizionale saranno i prodotti contenenti nuove sostanze attive non presenti in medicinali autorizzati in Europa alla data del 1 gennaio 2011, biologici e biosimilari, ma potranno essere inclusi anche i prodotti la cui autorizzazione è subordinata a particolari condizioni o autorizzati in circostanze eccezionali; i prodotti soggetti a studi sulla sicurezza dopo la concessione dell’AIC.
Questi medicinali sottoposti a monitoraggio addizionale saranno identificabili dal foglio illustrativo che recherà la dicitura “Medicinale sottoposto a monitoraggio addizionale” preceduta da un simbolo nero, il cui elenco sarà stilato dall’Agenzia Europea dei Medicinali.
La nuova normativa prevede la possibilità di imporre ai titolari di AIC, al momento della concessione della stessa o successivamente, di condurre ulteriori studi sulla sicurezza e/o sull’efficacia del farmaco.
Viene istituito all’interno dell’EMA il “Comitato di valutazione dei rischi per la farmacovigilanza“ (PRAC) in cui sono rappresentati tutti gli Stati membri.
Il PRAC copre tutti gli aspetti della gestione dei rischi derivanti dall’utilizzo dei medicinali per uso umano, anche per quanto riguarda l’individuazione, la valutazione, la riduzione e la comunicazione relativi al rischio di reazione avverse. Il PRAC dovrà fornire raccomandazioni al Comitato per i Medicinali per Uso Umano (CHMP) e al Gruppo di Coordinamento (CMD) su qualsiasi situazione emergente in farmacovigilanza e in relazione ai sistemi di gestione dei rischi monitorandone l’efficacia.
Infine la nuova legislazione fornisce disposizioni anche su procedure e/o tematiche specifiche inerenti le attività che le aziende farmaceutiche dovranno mettere in atto.
Il sistema previsto dalla nuova legislazione ed il suo funzionamento sono piuttosto complessi ed è necessaria una congrua dotazione di personale competente adeguatamente qualificato e addestrato, come specificato anche nel Regolamento di Esecuzione (UE) 520/2012 del 19 giugno 2012.
Le premesse per il raggiungimento di tali obiettivi sono state già poste in essere, mentre, per quanto riguarda i risultati bisognerà attendere il pieno funzionamento di quanto previsto, anche in termini di formazione del personale sanitario, con particolare riguardo alle attività di farmacovigilanza da parte delle strutture sanitarie e, più in generale, una maggiore partecipazione di tutte le parti interessate, inclusi i pazienti.
Indipendentemente dalle norme attualmente in vigore, le informazioni derivanti dall’esperienza professionale consentono di estendere la definizione di farmaco da banco a tutti i prodotti farmacologicamente attivi reperibili senza specifica prescrizione medica.
In questa definizione rientrano le seguenti categorie:
1) farmaci da banco propriamente detti;
2) farmaci senza obbligo di prescrizione medica;
3) farmaci soggetti a prescrizione medica, ma venduti senza presentazione di ricetta;
4) prodotti erboristici (Il problema della vigilanza sui prodotti erboristici è molto complesso).
Prendendo in considerazione i dati attualmente disponibili nella letteratura scientifica in merito ai criteri per l’autorizzazione alla vendita di un farmaco come prodotto da banco, è utile soffermarsi soprattutto su due aspetti: le indicazioni terapeutiche e il profilo di tollerabilità.
Per quanto riguarda le indicazioni terapeutiche, i farmaci da banco dovrebbero essere utilizzati solo per sintomi noti da tempo al paziente e per patologie che ne richiedano un uso limitato nel tempo. Inoltre tali patologie dovrebbero essere caratterizzate da una prognosi favorevole ed essere associate a un basso rischio di complicanze.
Per quanto riguarda invece il profilo di tollerabilità, i farmaci da banco dovrebbero possedere le seguenti caratteristiche:
- effetti avversi non gravi e reversibili;
- ampio margine terapeutico, con basso rischio di reazioni avverse gravi in caso di sovradosaggio;
- scarsa capacità di promuovere reazioni avverse o fallimenti terapeutici in seguito ad interazioni con altri farmaci;
- spettro di effetti avversi ben noti, facilmente diagnosticabili, e prontamente regredibili in seguito a un’interruzione della terapia (Lowe e Ryan-Wenger, 1999).
E’ utile ricordare che la maggior parte dei farmaci utilizzati per l’automedicazione rientra in una delle seguenti categorie:
- analgesici, antipiretici;
- farmaci attivi sul sistema respiratorio (anti-tosse, decongestionanti, anti-allergici);
- farmaci attivi sul tratto gastrointestinale (antiacidi, antisecretivi gastrici, antiemetici, procinetici, lassativi, antidiarroici);
- farmaci attivi sul metabolismo (integratori minerali, vitamine, prodotti per il controllo del peso corporeo).
Necessità di una vigilanza sui farmaci da banco
La necessità di svolgere un’attenta attività di vigilanza sui farmaci da banco deriva da considerazioni di ordine sia socio-economico che clinico-farmacologico. Dal punto di vista socio-economico, c’è una crescente tendenza da parte delle autorità sanitarie di vari Paesi a incoraggiare il ricorso all’automedicazione al fine di ridurre la spesa sanitaria. A questa spinta corrisponde una crescente disponibilità da parte delle autorità regolatorie a concedere autorizzazioni per la vendita dei farmaci come prodotti da banco. Il successo di queste iniziative è inoltre sostenuto da campagne promozionali intensive promosse dalle aziende farmaceutiche e dal fatto che un elevato numero di pazienti ricorre all’automedicazione a causa del costo eccessivo o della ridotta disponibilità di alcuni farmaci soggetti a prescrizione medica (Honig e Gillespie, 1998).
Per quanto riguarda gli aspetti clinico-farmacologici, è opportuno sottolineare che, sebbene i criteri proposti per l’autorizzazione alla vendita dei farmaci da banco siano rassicuranti, l’assunzione di questi prodotti può comportare un aumento del rischio di reazioni avverse gravi causate da un uso improprio o da abuso (Hughes et al., 1999). Infatti i farmaci da banco vengono assunti in assenza di controlli medici, ed è quindi possibile che i pazienti utilizzino tali prodotti per patologie non indicate, a dosi eccessive, o per periodi di tempo eccessivamente prolungati. I pericoli maggiori possono derivare dall’assunzione dei farmaci da banco in concomitanza con altri farmaci prescritti dal medico o con particolari alimenti, o con stati patologici che ne possono modificare in maniera significativa il profilo di efficacia o di sicurezza.
Interazioni tra farmaci
I rischi connessi alle interazioni tra prodotti da banco e farmaci soggetti a prescrizione medica non sono dissimili da quelli derivanti dalle interazioni tra farmaci prescrivibili. Infatti, l’assunzione di alcuni farmaci da banco, quali antiacidi, antagonisti dei recettori H2 dell’istamina, salicilati e altri farmaci anti-infiammatori non steroidei (FANS), prodotti anti-tosse, anti-raffreddore, anti-allergici, etc., può esporre il paziente al pericolo di interazioni sfavorevoli con altri farmaci prescritti dal medico (Honig e Gillespie, 1998; Sihvo et al., 2000).
Tra i farmaci da banco maggiormente utilizzati, i FANS possono dare luogo a vari tipi di interazioni farmacodinamiche. Per esempio, i FANS possono determinare una riduzione dell’effetto anti-ipertensivo in pazienti con ipertensione arteriosa in trattamento con beta-bloccanti, ACE-inibitori o diuretici. Ciò dipende dal fatto che i FANS, attraverso il blocco delle ciclo-ossigenasi, interferiscono negativamente con la sintesi di prostaglandine a livello renale. Tramite questo meccanismo i FANS possono causare un fallimento terapeutico in pazienti ipertesi sottoposti a trattamento con farmaci anti-ipertensivi. Un altro esempio è rappresentato dall’interazione dei FANS con la warfarina. Infatti alcuni FANS sommano la loro azione antiaggregante piastrinica a quella anticoagulante della warfarina determinando un aumento del rischio di reazioni avverse di tipo emorragico (Sihvo et al, 2000).
In molti casi i farmaci da banco causano interazioni di tipo farmacocinetico (Honig e Gillespie, 1998). Un esempio è rappresentato dagli antiacidi o dagli anti-H2 che attraverso un incremento del pH gastrico possono interferire con l’assorbimento di alcuni farmaci, quali chinoloni, profarmaci beta-lattamici o chetoconazolo. Inoltre, l’anti-H2 cimetidina può inibire la clearance epatica di vari farmaci, quali benzodiazepine, warfarina, fenitonina e alcuni antiaritmici. Altri esempi sono riportati nella tabella successiva.
Al rischio di interazioni sfavorevoli tra farmaci da banco e farmaci soggetti a prescrizione possono contribuire anche l’atteggiamento del paziente e del medico. Spesso il paziente non considera i prodotti da banco come veri e propri farmaci e non si preoccupa di riferirne l’uso al medico curante. Questi, d’altra parte, può prescrivere la propria terapia farmacologica senza interrogare il paziente sull’eventuale uso di farmaci da banco, contribuendo a determinare associazioni farmacologiche responsabili di reazioni avverse o fallimenti terapeutici.
Chiarimenti AIFA in merito ad alcuni termini
- Uso off-label: si riferisce a impieghi del medicinale usato intenzionalmente per finalità mediche non in accordo con le indicazioni di impiego autorizzate
- Misuso: si riferisce a situazioni in cui il medicinale è usato intenzionalmente ed in modo inappropriato non in accordo con le indicazioni di impiego autorizzate
- Abuso: si riferisce ad un intenzionale uso eccessivo del medicinale, sporadico o persistente, accompagnato da effetti dannosi fisici o psicologici
- Esposizione occupazionale: si riferisce all’esposizione ad un medicina come risultato di un impiego professionale o non professionale.
Attenzione alla «cascata» prescrittiva
La consapevolezza che la combinazione di numerose terapie è in grado di innescare una cascata prescrittiva può suggerire strategie più prudenti:
- Rivalutare attentamente la necessità del farmaco in causa.
- Utilizzare terapie non farmacologiche per gestire il problema.
- Ridurre la dose del farmaco in causa alla dose minima efficace.
- Considerare alternative farmacologiche più sicure in termini di rischio di reazioni avverse negli anziani.
Domande utili2
- È stata iniziata una nuova terapia o assunto un farmaco da banco o un integratore?
- Prima di cominciare una terapia farmacologica per curare un nuovo sintomo, considerare se si può trattare di una reazione avversa.
- La terapia iniziale, che ha portato alla cascata prescrittiva è veramente necessaria?
- Può essere sostituita con un’alternativa più sicura o può esserne ridotta la dose?
- Quali sono rischi e benefici della terapia che ha portato alla cascata prescrittiva?
- Rischi e benefici vanno considerati con il paziente condividendo le decisioni.
| Esempi di interazioni farmacocinetiche tra farmaci da banco (OTC) e farmaci soggetti a prescrizione medica. | ||
| Farmaco OTC | Farmaco prescritto | Risposta |
| Antiacidi | Chinoloni | Riduzione dell’assorbimento |
| Profarmaci beta-lattamici | Riduzione della biodisponibilità | |
| H2-antagonisti (cimetidina) | Chetoconazolo | Riduzione dell’assorbimento |
| Benzodiazepine | Inibizione della biotrasformazione | |
| Warfarina | Riduzione della clearance | |
| Flecainide, procainamide, chinidina, lidocaina, fenitoina | Riduzione della clearance | |
| FANS | Acido valproico | Spiazzamento dal legame proteico; inibizione della biotrasformazione (aspirina) |
| Litio | Aumento della concentrazione plasmatica (ibuprofene, naprossene) | |
| Digossina | Riduzione della clearance renale | |
| Metotressato | Potenziamento della tossicità del metotressato | |
Farmaci da banco e reazioni avverse: grado di consapevolezza del paziente
Alcuni studi hanno cercato di determinare quale sia il grado di consapevolezza dei pazienti in merito ai rischi potenzialmente connessi all’automedicazione con farmaci da banco. Hughes et al. (2002), dopo aver esaminato le abitudini di un vasto gruppo di pazienti per mezzo di specifici questionari, hanno osservato che la maggior parte dei soggetti mostra uno scarso grado di conoscenza dei possibili effetti avversi derivanti dall’uso di farmaci da banco. Nel caso dei farmaci soggetti a prescrizione, il paziente si preoccupa quasi sempre di interrogare il medico circa i rischi associati al trattamento e legge con attenzione le informazioni riportate nel foglietto illustrativo. Il prodotto da banco non viene invece considerato come un farmaco vero e proprio per cui il paziente tende a non acquisire informazioni sui rischi connessi alla sua assunzione. Lo studio ha inoltre evidenziato che i consumatori di farmaci da banco tendono a recepire meglio le informazioni sulle reazioni avverse fornite da personale sanitario qualificato, rispetto a quelle riportate nel foglietto illustrativo. Poiché non è il medico a guidare la scelta e le modalità di impiego dei farmaci da banco, è chiaro che il professionista qualificato a cui spetta l’obbligo di fornire le necessarie informazioni al paziente è rappresentato dal farmacista (Hughes et al., 2002).
Bibliografia
- Inman WHW, Weber JCP. The United Kingdom. In: Monitoring for Drug Safety. Inman WHW (Ed.). MTP Press, Lancaster, 1986; pp. 13-47.
- D’Arcy PF, Harron DWG. Background to the Conference. In: Proceedings of the Third International Conference on Harmonisation Yokohama. Queen’s University, Belfast, 1995; pp. 1-14.
- Edwards IR, Lindquist M, Wiholm B-E, Napke E, Quality criteria for early signals of possible adverse drug reactions. Lancet 1990; 336: 156-158.
- Bagaud B, Moride Y, Tubert-Bitter P, Chaslerie A, Haramburu F. False-positives in spontaneous reporting: should we worry about them? Br J Clin Pharmacol. 1994; 38: 401-404.
- Waller PC, Lee EH. Responding to drug safety issues. Pharmacoepidemiol Drug Saf 1999; 8: 535-552.
- Evans SJW, Waller PC, Davis S. Proposal reporting ratios: the uses of epidemiological methods for signal generation. Pharmacoepidemiol Drug Saf 1998; 7 (Suppl. 2): S102..
- Meyboom RHB, Hekster YA, Egberts ACG, Gribnau FWJ, Edwards IR. Causal or casual? The role of causality assessment in pharmacovigilance. Drug Saf 1997; 17: 374-389.
- Von Kries R. Neonatal vitamin K prophylaxis: the Gordian knot still awaits untying. BMJ 1998; 316: 161-162.
- Lees AJ on behalf of the Parkinson’s Disease Research Group in the United Kingdom. Comparison of therapeutic effects and mortality data of levodopa and levodopa combined with selegiline in patients with early, mild Parkinson’s disease. BMJ 1995; 311: 1602-1607.
- Hill AB. The environment and disease: association or causation? Proc Roy Soc Med 1965; 58: 295-300.
- Berlin JA. The use of meta-analysis in pharmacopeidemiology. In Pharmacoepidemiology, 2nd edn. Strom BL (Ed.). John Wiley, Chichester, 1994; pp. 525-547.
- Beardon PHG, McGilchrist MM, McKendrick AD, McDevitt DG, MacDonald TM. Primary non compliance with prescribed medication in primary care. BMJ 1993; 307: 846-848.
- Dackett DL, Hayes RB, Gent M, Taylor DW. Compliance. In: Monitoring for Drug safety. Inman WHW (Ed.). MTP Press, Lancaster, 1986; pp. 471-483.
- Evans JMM, MacDonald TM. Misclassification and selection bias in case-control studies using an automated database. Pharmacopidemiol Drug Saf 1997; 6: 313-318.
- Prescrizione dei farmaci di Pagni Aldo e Manfredi Carlo, edizioni C.G. Torino, 2002.
- Bryant BG, Lombardi TP. Cold, cough and allergy products. In: Handbook of nonprescription drugs, 10th edition, American Pharmaceutical Association, p. 89-115, 1993.
- Clark D, Layton D, Shakir SA. Monitoring the safety of over the counter drugs. We need a better way than spontaneous reports. Br Med J 323, 706-707, 2001.
- Corey JP, Houser SM, Ng BA. Nasal congestion: a review of its etiology, evaluation, and treatment. Ear Nose Throat J 79, 690-693, 2000.
- Honig PK, Gillespie BK. Clinical significance of pharmacokinetic drug interactions with over-the-counter (OTC) drugs. Clin Pharmacokinet 35, 167-171, 1998.
- Hughes GF, McElnay JC, Hughes CM, McKenna P. Abuse/misuse of non-prescription drugs. Pharm World Sci 21, 251-255, 1999.
- Hughes L, Whittlesea C, Luscombe D. Patients’ knowledge and perceptions of the side-effects of OTC medication. J Clin Pharm Ther 27, 243-248, 2002.
- Jones A. Over-the-counter analgesics: a toxicologic perspective. Am J Ther 9, 245-257, 2002.
- Kotecki JE. Factors related to pharmacists’ over-the-counter recommendations. J Community Health 27, 291-306, 2002.
- Krishnan HS, Schaefer M. Evaluation of the impact of pharmacist’s advice giving on the outcomes of self-medication in patients suffering from dyspepsia. Pharm World Sci 22, 102-108, 2000.
- Lowe NK, Ryan-Wenger NM. Over-the-counter medications and self-care. Nurse Pract 24, 34-44, 1999.
- Muller-Lissner S. Classification, pharmacology, and side-effects of common laxatives. Ital J Gastroenterol Hepatol 31 (Suppl. 3), S234-237, 1999.
- Sihvo S, Klaukka T, Martikainen J, Hemminki E. Frequency of daily over-the-counter drug use and potential clinically significant over-the-counter-prescription drug interactions in the Finnish adult population. Eur J Clin Pharmacol 56, 495-499, 2000.
- Stadlmann S, Zoller H, Vogel W, Offner FA. COX-2 inhibitor (nimesulide) induced acute liver failure. Virchows Arch 440, 553-555, 2002.
- Thomas J, Straus WL, Bloom BS. Over-the-counter nonsteroidal anti-inflammatory drugs and risk of gastrointestinal symptoms. Am J Gastroenterol 97, 2215-2219, 2002.
- Van Steenbergen W, Peeters P, De Bondt J, Staessen D, Buscher H, Laporta T, Roskams T, Desmet V. Nimesulide-induced acute hepatitis: evidence from six cases. J Hepatol 29, 135-141, 1998.
Tabella I. Numero di casi necessario per generare un segnale, se la frequenza dell’evento è rara nella popolazione generale (5)

Tabella II. Percentuale di reazioni avverse di un farmaco in base agli organi interessati
| Organo od apparato | % di ADR |
| Cute | 24 |
| Sintomi e segni generali | 21 |
| Sistema nervoso centrale | 17 |
| Apparato gastroenterico | 11 |
| Apparato respiratorio | 6 |
| Apparato muscolo scheletrico | 5 |
| Apparato cardiovascolare | 4 |
| Vari | 12 |
Tabella III. Uso del rapporto proporzionale di segnalazione (PRRs) per la generazione del segnale di allarme (5).

Tabella IV. Fattori da prendere in considerazione per ciascuna segnalazione che si riferisce a quel dato farmaco.
| Causalità | Valutazione della congruità temporale fra evento avverso e farmaco, effetto del dechallange e del rechallange, possibili cause alternative che spiegano l’evento. |
| Documentazione | Quanto più dettagliata è l’informazione, tanto più utile e valutabile essa sarà |
| Frequenza della segnalazione | Il numero di ADR sospettate in relazione all’uso del farmaco |
| Gravità della reazione | Quanto più grave essa è, tanto più importante sarà ai fini della salute pubblica. |
| Potenzialità del rischio | Se l’evento avverso dovesse essere frequente e dovuto ad un farmaco di uso molto ampio nella popolazione il rischio che la stessa vada incontro ad ADR sarà elevato e le implicazioni sanitarie di ciò saranno diverse da quelle da prendere a fronte di una ADR rara ed indotta da un farmaco poco usato dalla popolazione. Dovrà anche essere presa in considerazione l’evenienza che esistano già sul mercato altri farmaci della stessa classe o che il farmaco in questione sia realmente importante per la patologia per cui ne è stata approvata l’utilizzazione. |
Tabella V. Evidenze da considerare per valutare in maniera più approfondita il segnale che proviene da una serie di segnalazioni che si riferiscono a quel dato farmaco.
| Meccanismo d’azione del farmaco | Il farmaco potrebbe agire con un meccanismo che potrebbe spiegare l’insorgenza dell’evento avverso |
| Possibile effetto di classe | L’evento avverso è già stato riconosciuto per altri farmaci della stessa classe |
| Studi preclinici | Potrebbero aver messo in evidenza che il farmaco è metabolizzato da alcuni sistemi enzimatici soggetti ad inibizione (citocromo P40) e pertanto la contemporanea assunzione di altri farmaci potrebbe spiegare il perché dell’evento avverso. |
| Trials clinici | L’evento avverso potrebbe essere già stato osservato in questi studi, ma senza rilevare la sua incidenza, dato la limitata popolazione oggetto di tali studi. |
| Studi epidemiologici | Esistono studi che potrebbero essere stati condotti o che sono in corso atti a verificare se il farmaco in questione ha modificato l’incidenza dell’evento avverso sospettato. |
Tabella VI. Possibili spiegazioni alternative da considerare prima di imputare un evento avverso ad un determinato farmaco.
| Alternative farmacologiche | |
| Contemporanea assunzione di altri farmaci | Va esclusa la possibilità che l’evento avverso sia imputabile ad uno degli altri farmaci assunti. Esistono farmaci diversi che possono dare lo stesso evento avverso. |
| Interazioni tra farmaci | Il farmaco sospettato potrebbe aumentare la concentrazione ematica (e quindi la tossicità) di un altro farmaco. L’evento avverso pertanto andrebbe imputato al secondo farmaco e la spiegazione di esso sarebbe l’interazione. |
| Alternative non farmacologiche | |
| Presenza di patologia che crea confusione. | In presenza di una glomerulonefrite che si aggrava e che venga trattata con un aminoglicoside (che è noto avere potenzialità nefrotossiche) l’evento avverso è imputabile ala tossicità dell’aminoglicoside o al fatto che l’antibiotico non risulta efficace e la patologia si sta aggravando? |
| Insorgenza di un’altra patologia. | Durante un trattamento farmacologico per, ad esempio, una patologia ipertensiva, se dovesse insorgere un episodio di broncocostrizione andrebbe esclusa che questo fatto non sia imputabile a fattori non farmacologici. |
Tabella VII. Fattori che possono influenzare la possibilità di prevenire una ADR
| Caratteristiche del paziente | Caratteristiche del farmaco |
| Età | Via di somministrazione |
| Sesso | Formulazione (eccipienti, ecc.) |
| Razza | Regime posologico |
| Fattori genetici (polimorfismo) | Meccanismi di metabolizzazione |
| Malattie concomitanti (insufficienza epatica o renale | Via di escrezione |
| Compliance | Potenzialità di interazioni farmacologiche |
1 Fonte da sito AIFA https://www.aifa.gov.it/normativa-di-riferimento-farmacovigilanza
2 Aging: Is your patient taking too many pills? J Fam Pract. 2012 Nov;61(11):652-61.
Optimising drug treatment for elderly people: the prescribing cascade BMJ 1997;315:1096

